[box type=”bio”] What to Learn from this Article?[/box]
Early muscle weakness may be an initial manifestation of osteogenesis imperfect.
Case Report | Volume 7 | Issue 3 | JOCR May – June 2017 | Page 63-66| Vito Pavone, Teresa Mattina, Piero Pavone, Raffaele Falsaperla, Gianluca Testa. DOI: 10.13107/jocr.2250-0685.808
Authors: Vito Pavone[1], Teresa Mattina[2], Piero Pavone[3], Raffaele Falsaperla[3], Gianluca Testa[1]
[1] Department of Orthopaedics and Traumatologic Surgery, Ospedaliero – Universitaria Policlinico-Vittorio Emanuele, University of Catania, Catania, Italy.
[2] Department of Clinical and Experimental Sciences, University of Catania, Catania, Italy.
[3] Unit of Pediatrics and Pediatric Emergency, University Hospital Policlinico-Vittorio Emanuele, Catania, Italy.
Address of Correspondence:
Dr. Vito Pavone,
Ospedaliero – Universitaria Policlinico-Vittorio Emanuele, Via Plebiscito 628, 95100 Catania, Italy.
E-mail: vitopavone@hotmail.com
Abstract
Introduction: Osteogenesis imperfect (OI) is a heterogeneous and complex connective tissue disorder that manifests with low bone density and fragility. More than 15 types of OI have been distinguished on a clinical and molecular basis, but the classical clinical classification previously proposed in Types 1-4 with the recent inclusion of Type 5 appears to be more suitable. The diagnosis is mainly made on clinical and radiographic findings with fractures caused by mild trauma, bowing deformities of long bones, and growth deficiency. Non-skeletal features of the disorder include blue sclerae, hearing loss, decreased pulmonary function, cardiac valvular regurgitation, and muscle weakness.
Case Report: We report on a toddler girl affected by OI Type 1 who suffered from marked muscle weakness as the first initial sign, which led us to follow the diagnostic checklist for hypotonic children. The typical signs of the disorder later became evident and consistent with this diagnosis, including bone fractures and blue sclerae.
Conclusion: Early muscle weakness, previously unreported sign, may be an initial manifestation of OI, to be included in the differential diagnosis of disorders that cause hypotonia in childhood.
Keywords: Osteogenesis imperfecta, COLI AI gene, infantile hypotonia, motor delay, muscle involvement.
Introduction
Many factors make the diagnosis of motor delay in very young children particularly difficult. The diagnostic course of hypotonia at this age is hindered by difficulties in identifying the numerous etiologic events, the variability of the muscular tonus degree in childhood development, and the limited role of the diagnostic procedures at an early age [1, 2]. Recently, we examined a female toddler with a marked motor delay that mainly affected the lower limbs but with normal patellar reflexes, upper limb function, truncal control, and cognitive development. At the age of 13 months, the motor delay was so marked that muscle disorders were considered. The diagnosis of osteogenesis imperfecta (OI) was reached at the age of 28 months when the signs of the causal disorder occurred with the appearance of blue sclerae and undisplaced bone fractures. Molecular analysis subsequently confirmed the diagnosis of OI Type 1 by individuation of the autosomal mutation of the COLI A1 gene. The aim of this clinical report is to emphasize the role of motor delay as an initial, remarkable, and unreported manifestation of OI Type 1, to discuss the possible pathogenic causes and to report this sign in the differential diagnosis of other disorders presenting childhood hypotonia.
Case Report
A 13-month-old female complaining of marked motor delay was admitted to the University Hospital “Policlinico – Vittorio Emanuele” of Catania, Italy. She has a healthy 3-year-old sister. The patient’s parents are unrelated. The father was 35 years old and healthy. No bone fractures were reported for the father or other members of the paternal line. The mother was 31 years old, short in stature (147 cm height), and had no clear episodes of fractures. The maternal grandmother had suffered from various fractures as a result of mild trauma, she was also short in stature, and she died at the age of 50 due to vesical neoplasia. The grandmother’s sister was also short in stature and complained of two forearm fractures caused by mild traumatic events in adulthood. The mother reported not having had any infectious diseases or consuming alcohol or drugs during pregnancy, which were conducted normally. She recorded feeling normal fetal movements. The intrauterine ultrasound did not display any fetal anomalies.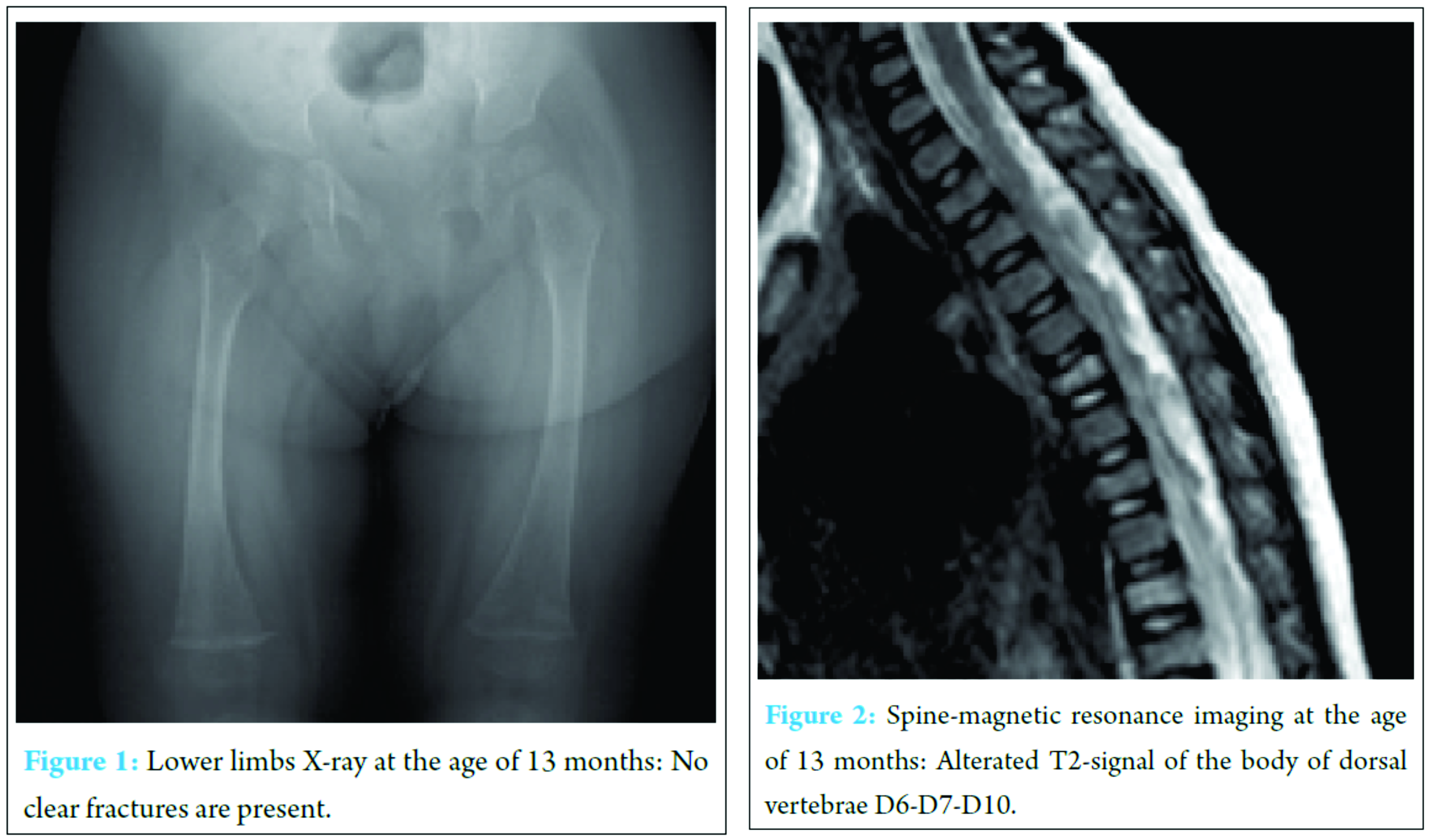
The patient was born by cesarean section with polyhydramnios at the 39th week of gestation with a birth weight of 2800 g, length of 48 cm, and head circumference of 35 cm. No perinatal problems were reported, and APGAR scores were 9 and 10 at 1 and 5 min, respectively. The development milestones were delayed, and the child had difficulty in maintaining a sitting position at the age of 7 months and in holding a standing position at 14 months. At admission, the girl weighed 8 kg with a height of 70 cm and head circumference of 44 cm (all in the third percentile). No malformative anomalies were evident, and no blue sclerae were observed. She interacted well with her parents and other people and showed good cognitive development. Arm and hand movements were possible, but she showed marked motor delay, particularly in the lower body. The patellar tendon reflexes, muscle strength, upper limbs function, and truncal control were not involved. The heart and respiratory system were normal as were the liver and spleen. Routine laboratory analysis was normal, including hemogram, coagulation testing, blood lactate, pyruvate, glucose and ketones, CK, copper and ceruloplasmine, plasma and urine amino acids, and urinary organic acid. Serum calcium levels were slightly elevated: 10.8 mg/dl (n.v. 8.8-10.6); phosphate: 5.0 mg/dl (n.v. 2.5-4.5). Electrocardiography, echocardiogram, wake and sleep electroencephalography, and electromyogram were normal. No bone lesions were found in lower limb X-ray (Fig. 1). Brain magnetic resonance imaging (MRI) was normal, and spine MRI showed signal alterations in dorsal vertebrae bodies D6, D7, and D10 (Fig. 2). The lower limb hypotonia persisted without a diagnosis, and physiotherapeutic treatment was initiated, which resulted in a slight improvement. At the age of 19 months, the parents opted for other medical consultations, in which a new spine MRI revealed a major involvement of the previously lesions in vertebral bodies D6-D7 and D10 with new vertebral involvements localized in the L2-L3 lumbar vertebrae. At the age between 22 and 25 months, the little girl showed various mild trauma fractures, the first localized on the right ulna, and subsequently, on the right tibia and fibula with early healing. We observed her again at the age of 28 months. The clear evidence of blue sclerae along with the easy fractures led us to the diagnosis of OI. The molecular analysis mutation of the COLI A1 gene displayed the germinal deletion c.52delC (p.<leu1 8Serfs*2) with subsequent FRAMESHIFT heterozygous at exone 1 level confirming the diagnosis of OI Type 1. The girl is now 3 years old, and her weight (11 kg), height (86 cm), and head circumference (49 cm) are all at lower limit. Cognitive development is good with no cranial nerve involvement, and patellar tendon reflexes are present. Lower limb hypotonia is still present but less marked than previously, not involving the upper limbs. The child is able to walk with mild limitation in running. No other motor dysfunctions were noticed. At the last physical examination, she showed mild articular hyperlaxity and asymmetry of the lower limbs (the left is longer 1 cm than the right). The anterior fontanel is still open (1 × 1 cm), and the dentinogenesis is normally developed. Laboratory examinations showed normal levels of serum erythrocyte sedimentation rate, C-reactive protein, alkaline phosphatase, lactate dehydrogenase, calcium, and phosphorus. Serum 25-hydroxyvitamin D was 26 ng/ml (n.v. 25-40). Thyroid evaluation, growth hormone, and parathyroid hormone were within normal levels. Treatment with diphosphonate was initiated.
Discussion
We began observing the patient at the age of 13 months due to a marked delay in motor development. She showed lower levels of growth and presented with hypotonia localized at the lower limbs, almost exclusively. No other clinical signs were reported: Cognitive development was good, no clear malformative anomalies were present, and patellar tendon reflexes were normal. The severe hypotonia led us to perform the diagnostic checklist for muscle disorders, which indicated no results for muscle involvement. A non-specific light bone alteration in vertebral D6, D7, and D10 was noticed. The mother also initially omitted the history of bone fragility in some members of her family. OI was diagnosed after the classical signs appeared (bone fractures and blue sclerae). The diagnosis was confirmed by molecular analysis, which indicated mutation of the COLI A1 gene. OI or brittle bone disease is a fairly uncommon, complex, and heterogeneous connective tissue disorder that manifests with low bone mass, bone fragility, recurrent bone fractures, and substantial linear growth deficiency [3, 4, 5]. The general incidence is calculated to be one in 15-20.000 newborns [2]. The initial diagnosis of OI is mainly based on clinical and radiographic findings of fractures from mild trauma. Sillence et al. [3] classified four types of OI according to their clinical and radiological features, mainly with autosomal dominant inheritance and ranging from mild to lethal. The recent molecular discoveries have widely expanded and distinguished the types of OI. There are five groups based on the metabolic pathway involved, including forms related to collagen synthesis, structure, and processing; post-translational modification; folding and cross-linking; mineralization; and osteoblast differentiation [2, 4]. Nowadays, the most current used is the clinical classification previously proposed by Sillence et al. [1, 3] with the recent inclusion of Type 5 with specific radiological features, consisting of interosseus membrane calcifications. OI is considered a predominantly collagen-related disorder [4]. Collagen is the main structure protein of the extracellular matrix present within various connective tissues, and it is widely expressed in the entire body, including the tendons, ligaments, skin, cornea, bones, vertebral discs, and dentine. Collagen constitutes 2-3% of muscle tissue and is one of the major components of the endomysium [2]. In all groups, according to the age and grade of severity, the disorder may manifest with skeletal features as well as with chest wall deformities and vertebral impairment. Non-skeletal features include blue sclerae, vascular fragility, progressive hearing loss, dentinogenesis anomaly, decreased pulmonary function, and cardiac valvular regurgitation [4]. OI Type 1 is distinctly milder and the most frequently encountered form, comprising about 85% of the cases. It is associated with mutations in collagen Type 1 alpha 1 (COLI A1) or alpha 2 (COLI A2) genes. Type 1 collagen is a heterotrimer containing two alpha 1 (1) and one alpha 2 (1) chains [6]. Heterozygous null COLI A1 alleles result in milder form of OI, whereas the homozygous null alpha 2 (1) mutations present with symptoms encompassing severe OI to cutis laxa, joint hyperlaxity type Ehlers-Danlos syndrome [2, 4]. According to Marini et al. [2], patients with COLI A1 show an autosomal dominant inheritance and clinically present with normal stature, little or no deformity, blue sclerae, and hearing loss in 50% of cases. Patients may have early osteoporosis and present their first fracture in the pre-school years, and the number of fractures tends to decrease in the post-pubertal period. DEXA z-scores are reported to range from −1 to −3. Dentinogenesis in these patients may (subtype 1) or may not (subtype 2) be involved [2, 3, 7, 8]. The bones in OI Type 1 are normally modeled, usually with no bowing of the long bones, and central vertebral compressions are uncommon [7, 8, 9]. Muscle weakness, joint laxity, fatigue, diminished exercise capacity, and limitations in overall strength during walking, running, and living activities have been reported [10, 11, 12, 13]. In contrast to what is usually reported in individuals with OI, the patient showed early and marked motor delay that was almost exclusively localized in the lower limbs. Weak muscle tone and joint laxity have been reported in patients with OI, even at a young age, but it has not been sufficiently evaluated among the features of this disorder. Handheld dynamometry performed on 17 children and adolescents affected by OI Type 1 showed muscle weakness compared to normal reference values [10]. In contrast, Caudill et al. [11] used the same system and found no differences in 20 children with OI Type 1 compared to 20 age-matched healthy controls. Veilleux et al. [12] assessed lower extremity dynamic muscle function in 54 patients (6-21 years old) with OI Type 1 and 54 age- and sex-matched controls. The muscle anatomy was measured through the cross-sectional area of the calf muscle, density was measured through peripheral quantitative computed tomography, and lower extremity muscle function was registered by jumping mechanography. The results showed smaller muscle size in the affected patients with a normal muscle density and a significant force deficit. In a sample of 31 individuals with OI Type 1, almost all acquired unsupported gait by the age of 18 months, while delayed gait acquisition was found in children who had fractures in the lower limbs and/or orthopedic surgery [13]. Functional muscle-bone units were studied in 30 children and adolescents with OI and matched controls by measuring tibia mineral content and peak force of the lower extremities. The results showed that the muscle-bone relationship was similar between groups. The authors speculated that muscle weakness may have acted in OI patients as a negative factor in decreasing bone strength [14]. According to Pouliot-Laforte et al. [15], it is not clear if the muscle weakness observed in some cases of OI Type 1 is due to the low physical activity of the patients or if it results from impaired collagen Type 1 synthesis in the muscles or tendons. In 14 children and matched controls, they found lower muscle force and power in the group of OI patients, although they were as active as their healthy counterparts. According to Bregou Bourgeois et al. [1], OI Type 1 children may present with muscle weakness, but this is not directly related to poor physical activity of the affected patients. In our patient, hypotonia was particularly notable and was initially the only presenting sign of the disorder and was localized mainly in the lower limbs. Only later did the diagnosis of OI Type 1 become clear based on the typical features represented by blue sclera and various bone fractures. Muscle weakness has been reported among the features of OI Type 1, but it is overshadowed by symptoms related to bone fractures and it is reported not so early and marked as in the present patient. The first spine MRI revealed in the patient a not specific flattening localized in the D6, D7, and D10 vertebral bodies, and the vertebral anomalies became more evident in the following examinations performed 1 year later. These vertebral bodies’ anomalies could have contributed to the hypotonia of the patient, but this hypothesis contrasts with the course of the muscle weakness, which improved partially after his physical activity. Another hypothesis is that like the other skeletal and non-skeletal features of OI, muscle involvement in the patient appears to be pathogenetically linked to generalized disorder in the connective pathway. In the patient, the mutation COLI A1, directly or in association with other factors, may have acted on the muscular collagen more markedly and more precociously, causing clear muscle weakness. To the best of our knowledge, this report is the first example of marked and precocious muscular involvement in OI Type 1 presenting before the onset of fractures.
Conclusion
Toddler hypotonia may be the initial sign of OI Type 1. This sign seems to be associated with connective-related disorder and should be considered in the differential diagnosis of children manifesting early hypotonia. Marked hypotonia, affecting almost exclusively the lower limbs, and progressive improvement of this clinical sign, before treatment with diphosphonate, remain to be explained.
Clinical Message
OI is a connective tissue disorder with low bone density and fragility. Early muscle weakness may be an initial manifestation of this disorder.
References
1. Bregou Bourgeois A, Aubry-Rozier B, Bonafé L, Laurent-Applegate L, Pioletti DP, Zambelli PY, et al. Osteogenesis imperfecta: from diagnosis and multidisciplinary treatment to future perspectives. Swiss Med Wkly 2016;146:w14322.
2. Marini JC, Reich A, Smith SM. Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Curr Opin Pediatr 2014;26(4):500-507.
3. Sillence DO, Barlow KK, Cole WG, Dietrich S, Garber AP, Rimoin DL, et al. Osteogenesis imperfecta Type III. Delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet 1986;23(3):821-832.
4. Forlino A, Marini JC. Osteogenesis imperfecta. Lancet 2016;387(10028):1657-1671.
5. Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015;167A(12):2869-2892.
6. Willing MC, Deschenes SP, Slayton RL, Roberts EJ. Premature chain termination is a unifying mechanism for COL1A1 null alleles in osteogenesis imperfecta Type I cell strains. Am J Hum Genet 1996;59(4):799-809.
7. Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 2014;164A(6):1470-1481.
8. Aubry-Rozier B, Unger S, Bregou A, Freymond Morisod M, Vaswani A, Scheider P, et al. News in osteogenesis imperfecta: from research to clinical management. Rev Med Suisse 2015;11(466):657-658, 660.
9. Calder AD. Radiology of osteogenesis imperfecta, rickets and other bony fragility states. Endocr Dev 2015;28:56-71.
10. Takken T, Terlingen HC, Helders PJ, Pruijs H, Van der Ent CK, Engelbert RH, et al. Cardiopulmonary fitness and muscle strength in patients with osteogenesis imperfecta Type I. J Pediatr 2004;145(6):813-818.
11. Caudill A, Flanagan A, Hassani S, Graf A, Bajorunaite R, Harris G, et al. Ankle strength and functional limitations in children and adolescents with Type I osteogenesis imperfecta. Pediatr Phys Ther 2010;22(3):288-295.
12. Veilleux LN, Lemay M, Pouliot-Laforte A, Cheung MS, Glorieux FH, Rauch F, et al. Muscle anatomy and dynamic muscle function in osteogenesis imperfecta Type I. J Clin Endocrinol Metab 2014;99(2):E356-E362.
13. Brizola E, Staub AL, Félix TM. Muscle strength, joint range of motion, and gait in children and adolescents with osteogenesis imperfecta. Pediatr Phys Ther 2014;26(2):245-252.
14. Veilleux LN, Pouliot-Laforte A, Lemay M, Cheung MS, Glorieux FH, Rauch F, et al. The functional muscle-bone unit in patients with osteogenesis imperfecta Type I. Bone 2015;79:52-57.
15. Pouliot-Laforte A, Veilleux LN, Rauch F, Lemay M. Physical activity in youth with osteogenesis imperfecta Type I. J Musculoskelet Neuronal Interact 2015;15(2):171-176.
 |
 |
 |
 |
| Dr. Vito Pavone | Dr. Piero Pavone | Dr. Raffaele Falsaperla | Dr. Gianluca Testa |
| How to Cite This Article: Pavone V, Mattina T, Pavone P, Falsaperla R, Testa G. Early Motor Delay: An Outstanding, Initial Sign of Osteogenesis Imperfecta Type 1. Journal of Orthopaedic Case Reports 2017 May-June;7(3):63-66. |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com