[box type=”bio”] Learning Point of the Article: [/box]
How to differentiate the potential differential diagnostic conditions before a diagnosis of chondromyxoid fibroma is made which is essentially a diagnosis of exclusion.
Case Report | Volume 9 | Issue 4 | JOCR July-August 2019 | Page 101-105 | G HemanthaKumar, Muthu Sathish. DOI: 10.13107/jocr.2019.v09i04.1500
Authors: G HemanthaKumar[1], Muthu Sathish[1]
[1]Department of Orthopaedics and Traumatology, Rajiv Gandhi Government General Hospital, Chennai, Tamil Nadu, India.
Address of Correspondence:
Dr. Muthu Sathish,
Department of Orthopaedics and Traumatology, Rajiv Gandhi Government General Hospital, Chennai – 600 003, Tamil Nadu, India.
E-mail: drsathishmuthu@gmail.com
Abstract
Introduction: Chondromyxoid fibroma (CMF) is a benign rare bone tumor of slow-growing nature arising from chondroblastic derivation. CMF in most of the cases is a diagnosis of exclusion, and in this case report, we differentiate the histological and radiological findings of CMF and difficulties in diagnosis of CMF from potential differential diagnosis.
Case Report: A 38-year-old female patient presented with a history of limping for 5 months and on evaluation revealed an expansile osteolytic lesion in fibular head with septations and soft tissue component. Excision biopsy was done. Histological examination revealed a cellular neoplasm arranged as vague nodules in chondroid background with occasional mitotic figures and giant cells in periphery without any calcification. To rule out chondroblastoma, S-100 and epithelial markers were done which was negative establishing diagnosis of CMF by exclusion.
Conclusion: CMF is often misdiagnosed being a radiological and pathological mimicker. Histology remains key to diagnosis. En bloc resection remains the mainstay of management in expendable bone-like fibula.
Keywords: Chondromyxoid fibroma, benign bone tumor, en block resection, fibula.
Introduction
Chondromyxoid fibroma (CMF) is a benign rare bone tumor of slow-growing nature arising from chondroblastic derivation [1, 2, 3, 4]. Jaffe and Lichtenstein described the condition at first in 1943 [5]. In their work, they differentiated CMF from chondrosarcoma, which is a much more common, but with malignant nature. Before their description in 1943, CMF has been considered as a giant cell variant or a benign cartilaginous tumor. A myxoid element in the tumor is the defining characteristic feature. Around 500 CMF cases have been described in the entire literature [6]. The diagnostic frequency seems to be in decline. CMF mainly affects the second and third decade of young adults. Around 80% of patients are <36 years. The tumor is not gender specific and both males and females are affected equally; however, some series showed a slight male predominance [7]. The aim of our study is to evaluate the histological and radiological findings of CMF and difficulties in diagnosis from potential differential diagnosis of CMF. Consent was obtained for publication of the case details without revealing the identity of the patient.
Case Report
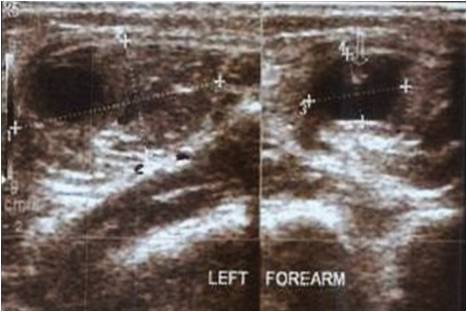
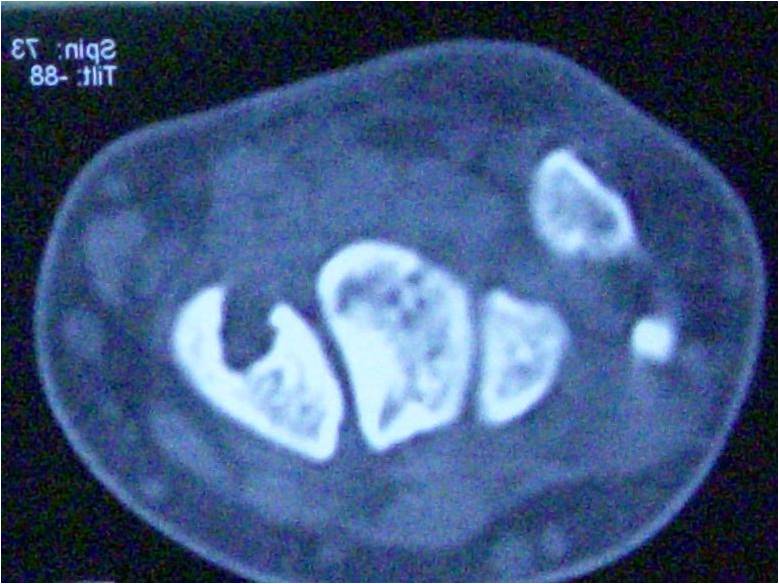
We present a case report of a 38-year-old female who presented with difficulty in walking for the past 5 months associated with pain in the knee for the past 3 months. The pain was dull aching in nature, mild intensity. There was no diurnal variation. Physical examination showed mild tenderness. Overlying skin was normal with restricted range of movement due to pain. Blood investigations were under normal limits. On evaluation radiologically, an expansile osteolytic lesion located in the head of the fibula with thinning and septations measuring 33 × 25 × 18 mm noted. Narrow zone of transition and no matrix mineralization noted. Cortex discontinuity noted in the lateral aspect. Joint space was normal and proximal tibial shaft appeared normal and soft tissue component was noted as shown in (Fig. 1).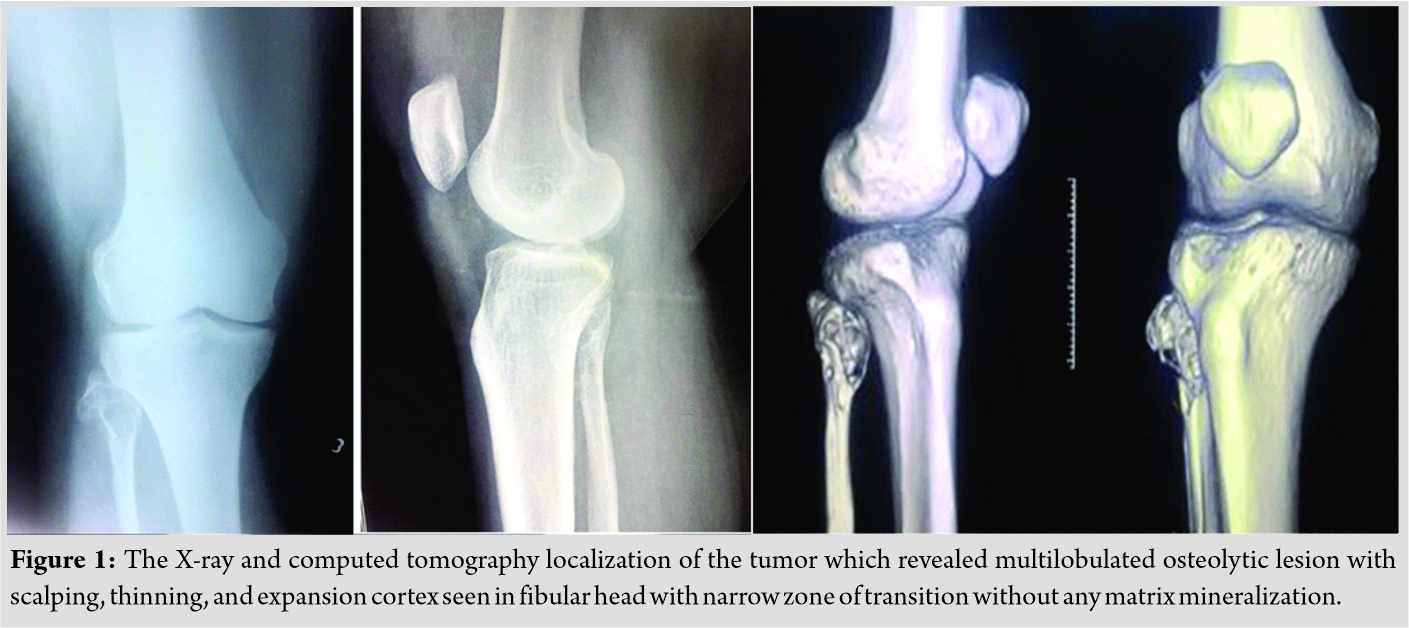
On magnetic resonance imaging, T1-T2 hypointense and short tau inversion recovery hyperintense lesion with thinning and endosteal scalloping of the cortex with focal break noted in the proximal fibula without any periosteal reaction as shown in (Fig. 2).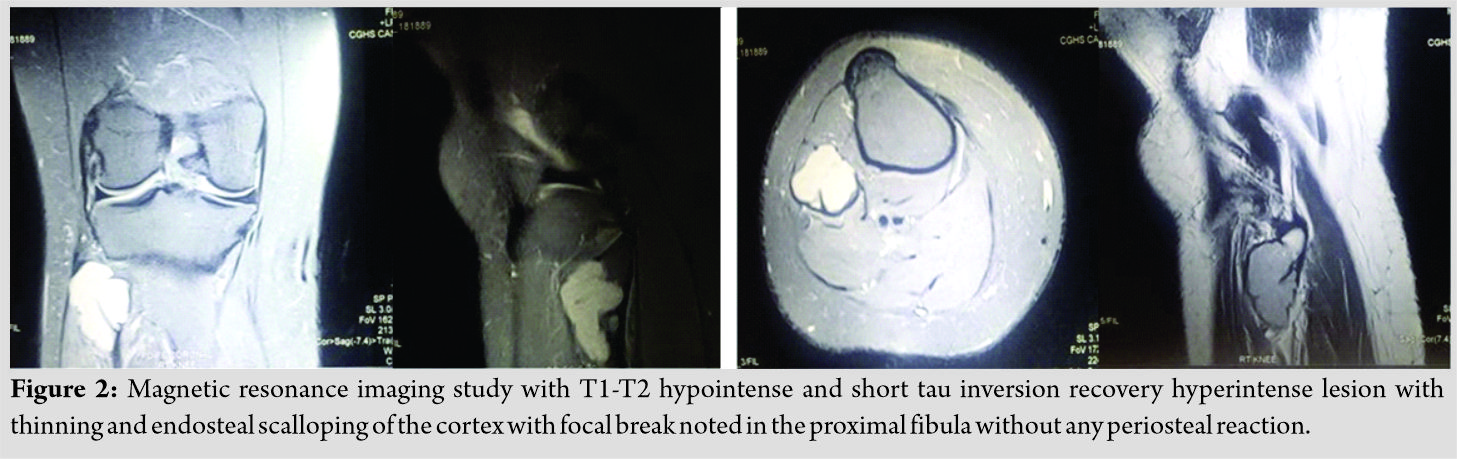
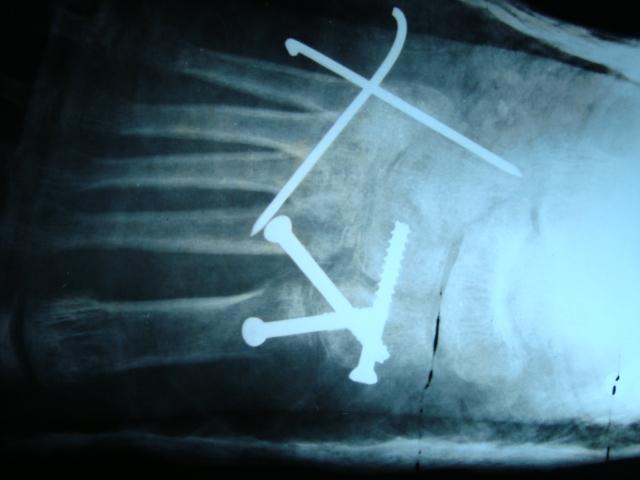
Excision biopsy was planned and after obtaining consent from the patient, an en block excision was done. The resected specimen was sent to histopathological examination. Gross examination showed a grayish-white ill-defined lesion at the epi-metaphyseal region measuring 2.5 × 1.5 × 1 cm which is focally extending into the adjacent soft tissue. On serial microscopic sectioning and examination as shown in (Fig. 3) revealed cellular neoplasm which was well demarcated from the adjacent osseous tissue arranged as vague nodules in chondroid background. The cells were round to oval with vesicular nuclei and abundant eosinophilic cytoplasm and displayed well-delineated cell borders. Mitosis is occasional 1–2/10 hpf.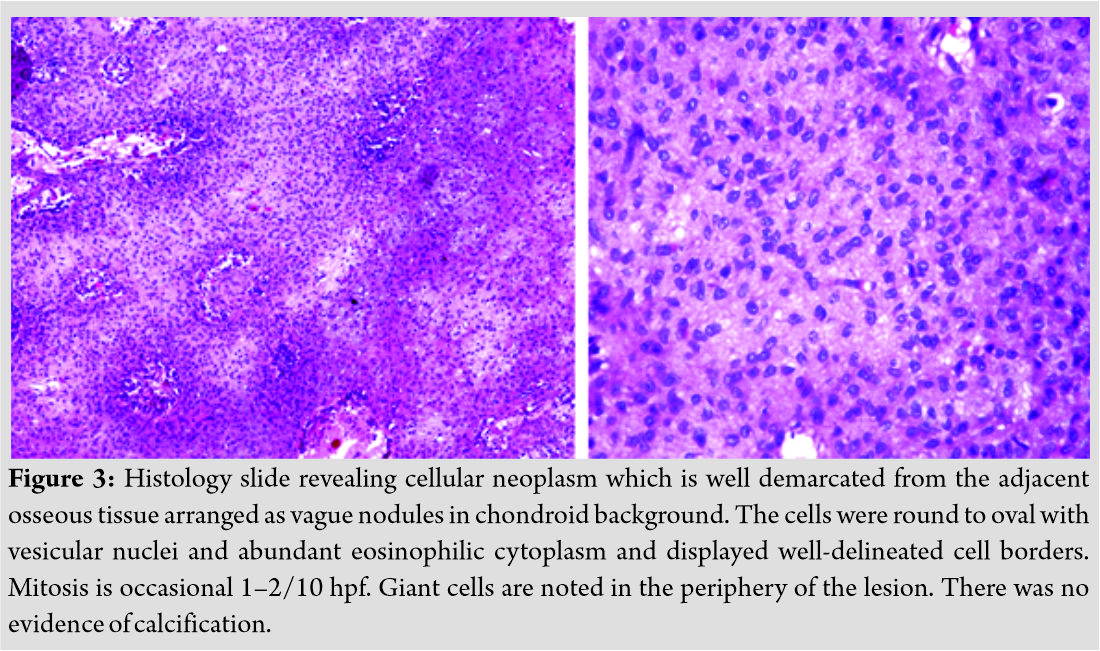
Giant cells are noted in the periphery of the lesion. There was no evidence of calcification. With a suspicion for chondroblastoma, S-100 and epithelial markers were done which all came out to be negative establishing the diagnosis of CMF by exclusion. Although myxoid element was not much evident on histology considering the tumor to be in the earlier stages of evolution diagnosed, early diagnosis of CMF was made. Postoperatively, the patient was put on partial weight-bearing for initial 2 weeks and later resumed full weight-bearing by 4 weeks. The case was followed up for 2 years without any recurrence till date as shown in (Fig. 4)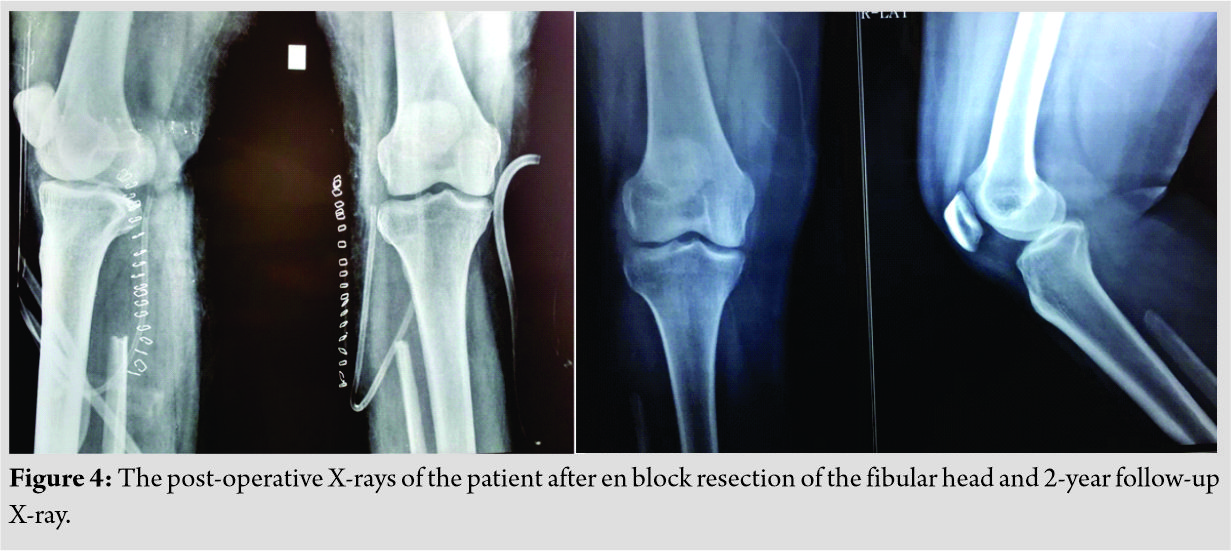
Discussion
The common location of CMF is metaphysis adjacent to the growth plate, which strengthens the theory that lesion is of cartilaginous remnant origin [4]. Similar reports of fibular CMF have been made by Mane et al. [8] from India and Atalar et al. [9] from Turkey and Merine et al. [10] from New York who also corroborate the rarity of the tumor incidence and location in fibula. To make a certain diagnosis, a thorough clinicoradiological and pathological examination was important as CMF is often misdiagnosed as chondroblastoma or chondrosarcoma due to some pathological similarities. A myxoid element is the defining characteristic of this tumor, and hence, histology remains the key in diagnosing this rare entity. However, the myxoid element was not so evident in our case which made us to diagnose this pathology by exclusion of the other possibilities such as chondroblastoma by special stains and epithelial markers. Since the tumor is in initial stages of differentiation, myxoid element may not be evident in our case. Grossly, the tumor is firm, grayish-white in color which may be lobulated or pseudolobulated. Their structure resembles that of fibrous tissue or hyaline cartilage as in tumors such as chondroblastoma or chondrosarcoma. The lesions often thin the cortex and rarely destroy trabecular bone. Rarely, the tumor has some areas of hemorrhagic degeneration and cystic degeneration and sometimes mimics aneurysmal bone cysts. Many CMFs exhibit morphological features that show to be in different stages of chondrogenesis [11, 12]. The most characteristic pathological fracture is the myxoid element. Giant cells are mostly seen toward the periphery of the tumor architecture. Low grade chondrosarcoma sometimes mimic CMF histologically, except for the lack of myxoid element and soft tissue involvement. Chondrosarcoma has peculiar demographic and radiographic features. Chondrosarcoma has peak incidence around the sixth and seventh decades of life, whereas CMF occurs in the second and third decade of life. Radiographically, location of the chondrosarcoma is central and has rich calcifications. Both chondrosarcoma and CMF sometimes show cortical expansion. In chondrosarcomas with long-standing or higher grade type, soft tissue involvement and cortical erosion are noted [13, 14]. CMF histopathological picture shows a lobular pattern of growth with focal hyaline deposits and rare mitotic figures and even cellular pleomorphism sometimes which may be similar to that of chondrosarcoma. However, chondrosarcoma shows features such as rich mucinous material, higher nuclear atypia, and multiple pleomorphic or multinuclear cartilage cells. Moreover, it behaves in a more malignant fashion. Patients with CMF may be misdiagnosed in the early stages as giant cell tumor (GCT) of bone, but they are much older than persons with CMF; moreover, cellular and radiographic features of GCT differ from that of CMF. GCTs are usually of metaphyseal origin with epiphyseal extension. Multinucleated giant cells with nucleus identical to the background nucleus of the stromal cells are the major defining histologic characteristic of GCT [15]. CMFs show only a few giant cells which are also located near the periphery. GCTs show neither myxoid nor chondroid elements. CMFs are managed surgically with intralesional curettage or en bloc excision depending on the location of the lesion [17]. Although Jaffe and Lichtenstein showed in their original description of CMF that incomplete removal did not lead to recurrence and the remnant tissue shows regression [5], some series noted recurrence rates of approximately 25% with curettage and bone grafting which may be higher in young children who are in the first or second decade of life and in myxoid predominant CMFs. Aggressive curettage of the tumor in the vicinity of the physis resulted in physeal damage many cause growth arrest. En bloc excision is usually curative in expendable bones. Recurrence is noted in cases, in which marginal excision was done. In a study by Bharma et al. of 22 consecutive cases of CMF treated by intralesional curettage [16], 2 patients (9%) showed local recurrence within 2 years after curettage. CMFs in axial skeleton may behave in an aggressive fashion after resection. Malignant conversion following en block resection is extremely rare. Consequently, no case of mortality was reported due to true CMF. Extended curettage with phenol, methyl methacrylate, and liquid nitrogen has not decreased the recurrence. In occurrence of this tumor in expendable bones such as fibula, en block resection is the ideal treatment of choice to prevent future recurrence. Radiotherapy may be considered an option for unresectable tumors [15]. Radiotherapy is generally avoided to prevent post-radiation sarcoma. However, malignant transformation in the absence of preceding radiotherapy has been noted which many authors believe could be a misdiagnosed chondrosarcoma. Recurrence if at all happens mostly within 2 years, but sometimes recurrence after 19 years has also been reported [13, 16, 17, 18]. Hence, routine patients follow-up with periodic physical and radiological examination for a minimum period of 2 years is recommended [19, 20].
Conclusion
CMF is a rare tumor of bone which is often confused with other radiological and pathological mimickers and misdiagnosed many times. Histological evaluation remains a key in diagnosing this rare pathology which is at times a diagnosis of exclusion as in our case. En bloc resection remains the mainstay of the management of this tumor in expendable bone-like fibula.
Clinical Message
CMF a rare tumor, is often confused with other radiological and pathological mimickers such as chondroblastoma or chondrosarcoma. However, myxoid element in histology is the defining characteristic feature. En bloc resection is curative for expendable bone-like fibula.
References
1. McGrory BJ, Inwards CY, McLeod RA, Sim FH. Chondromyxoid fibroma. Orthopedics 1995;18:307-10.
2. Ralph LL. Chondromyxoid fibroma of bone. J Bone Joint Surg Br 1962;44-B:7-24.
3. Schutt PG, Frost HM. Chondromyxoid fibroma. Clin Orthop Relat Res 1971;78:323-9.
4. White PG, Saunders L, Orr W, Friedman L. Chondromyxoid fibroma. Skeletal Radiol 1996;25:79-81.
5. Jaffe HL, Lichtenstein L. Chondromyxoid fibroma of bone; a distinctive benign tumor likely to be mistaken especially for chondrosarcoma. Arch Pathol (Chic) 1948;45:541-51.
6. Blackwell JB, Curnow MN. Benign bone tumours in Western Australia, 1972-1996. Pathology 2007;39:567-74.
7. Wu CT, Inwards CY, O’Laughlin S, Rock MG, Beabout JW, Unni KK, et al. Chondromyxoid fibroma of bone: A clinicopathologic review of 278 cases. Hum Pathol 1998;29:438-46.
8. Mane K, Singhania S, Khan SM, Gudhe M, Khan S, Singh P. Chondromyxoid fibroma of the distal one-third of the fibula: A rare tumour at rare site. Clin Transl Orthop 2016;1:126-7.
9. Atalar H, Başarir K, Uraş I, Yildiz Y, Erekul S, Sağlik Y, et al. Chondromyxoid fibroma: An evaluation of 11 patients. Acta Orthop Traumatol Turc 2007;41:31-5.
10. Merine D, Fishman EK, Rosengard A, Tolo V. Chondromyxoid fibroma of the fibula. J Pediatr Orthop 1989;9:468-71.
11. Romeo S, Hogendoorn PC, Dei Tos AP. Benign cartilaginous tumors of bone: From morphology to somatic and germ-line genetics. Adv Anat Pathol 2009;16:307-15.
12. Bhamra JS, Al-Khateeb H, Dhinsa BS, Gikas PD, Tirabosco R, Pollock RC, et al. Chondromyxoid fibroma management: A single institution experience of 22 cases. World J Surg Oncol 2014;12:283.
13. Dahlin DC. Chondromyxoid fibroma of bone, with emphasis on its morphological relationship to benign chondroblastoma. Cancer 1956;9:195-203.
14. Marin C, Gallego C, Manjón P, Martinez-Tello FJ. Juxtacortical chondromyxoid fibroma: Imaging findings in three cases and a review of the literature. Skeletal Radiol 1997;26:642-9.
15. Dürr HR, Lienemann A, Nerlich A, Stumpenhausen B, Refior HJ. Chondromyxoid fibroma of bone. Arch Orthop Trauma Surg 2000;120:42-7.
16. Heydemann J, Gillespie R, Mancer K. Soft tissue recurrence of chondromyxoid fibroma. J Pediatr Orthop 1985;5:725-7.
17. Hristov B, Shokek O, Frassica DA. The role of radiation treatment in the contemporary management of bone tumors. J Natl Compr Canc Netw 2007;5:456-66.
18. Gherlinzoni F, Rock M, Picci P. Chondromyxoid fibroma. The experience at the istituto ortopedico rizzoli. J Bone Joint Surg Am 1983;65:198-204.
19. Mikulowski P, Ostberg G. Recurrent chondromyxoid fibroma. Acta Orthop Scand 1971;42:385-90.
20. Zillmer DA, Dorfman HD. Chondromyxoid fibroma of bone: Thirty-six cases with clinicopathologic correlation. Hum Pathol 1989;20:952-64.
 |
 |
| Dr. G Hemantha Kumar | Dr. Muthu Sathish |
| How to Cite This Article: G HemanthaKumar, Sathish M. Diagnosis and Literature Review of Chondromyxoid Fibroma – A Pathological Puzzle. Journal of Orthopaedic Case Reports 2019 Jul-Aug; 9(4): 101-105. |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com