Perthes is a biological disease, its manifestations can mimic other conditions and it´s necessary to consider rare and treatable diseases in a child with limp or hip pain.
Dr. Douglas Colmenares-Bonilla, Department of Pediatric Orthopedic, Hospital Regional de Alta Especialidad del Bajio, Milenio 130, Col San Carlos La Roncha, Leon, Guanajuato, CP 376 70 Mexico. E-mail: douglas_cb@yahoo.com
Introduction:Perthes disease radiography can mask other diseases. Properly observing the X-ray and changes in other joints helps to discard Perthes-like conditions. Association with changes in other systems, as well as the pathological history and even the examination of family members can help diagnose early attenuated cases of rare diseases.
Case Presentation:The case of a girl with hip pain, limp and doubtful radiographs is presented. Perthes disease was diagnosed elsewhere and physical therapy and analgesics were prescribed. As a second opinion, family look for another orthopedic physician, who looks beyond hip pain and limp. Full body X-rays are suggestive of skeletal dysplasia. Metabolic disease diagnosis is suspected with pathological history of the younger brother, with repetitive respiratory infections, as well as abnormal X-ray panel. Multiple dysostoses in both patients are demonstrated. The above findings help to diagnose Morquio disease in an attenuated variant after molecular examinations.
Keywords: Perthes, skeletal dysplasia, attenuated morquio, pelvis, mucopolysaccharidoses.
Pelvis X-rays are a common study in the pediatric orthopedics’ practice. Of the changes observed in this study, they may suggest different diseases. Perthes disease shows changes secondary to necrosis in the femoral epiphysis, acetabular changes are rare, and other joint changes, null [1]; in skeletal dysplasia instead, bone changes are observed all over different joints. In metabolic diseases, bone skeletal changes are seen in conjunction with other non-skeletal manifestations [2].
Mucopolysaccharidosis IV A (MPS IV) also called Morquio syndrome, is a rare genetic disorder with autosomal recessive inheritance, is caused by total or partial deficiency of N-acetylgalactosamine-6-sulfate sulfatase enzyme. Consequently, an accumulation of glycosaminoglycans occurs in the lysosomes, interfering with the function of the cell. Its frequency is difficult to calculate due to high variability in the way it is reported in different countries or by different sources [3]. At birth, it ranges from 1/71,000 to 1/179,000 live borns (3, 4). It has been described as a disease with a broad spectrum of phenotypes, diagnosed in all ethnic groups. Its early detection is often hampered by broad variability in its manifestations. Typical cases usually take about 2 years to make the diagnosis after the onset of symptoms, but mild cases may be undiagnosed until adult age [4, 5, 6, 7].
Morquio’s disease manifests as a progressive skeletal dysplasia with disproportionate growth-producing multiple bone and joint deformities, airway obstruction, pulmonary restriction, and multiple damages in other systems. Without adequate treatment, these alterations have a negative impact on patients’ daily life [6]. Although skeletal alterations are present in all patients, they do not always produce obvious clinical manifestations at early age or are not so evident. On the other hand, bone alterations can be confused with localized orthopedic conditions, such as Perthes disease or primary skeletal dysplasia such as spondyloepiphyseal dysplasia [8]. The aim of this report is to show lysosomal deposit etiology in two siblings whose main skeletal complaint initially suspected Perthes’ disease.
It is a couple of brothers, male and female, 7 and 8 years old, respectively.
The 7-year-old female starts 3 months before her assessment with a right hip limp and mild pain worsened with physical activity (3/10 in a visual analogous scale of pain). There was no trauma history.
Previously examined elsewhere, she is diagnosed as suffering bilateral Perthes. Physical therapy, analgesics, and activity restrictions are prescribed (Fig. 1).
Consanguinity, family history of orthopedic or deposit diseases are denied. She is the third and last pregnancy of a 35-year-old mother, with a normal course and full-term regular delivery. No perinatal considerations are reported.
Surgery at 1 month of life because of urachal canal persistency.
At physical examination, she is a patient weighing 24.3 kg (50th percentile, using the World Health Organization standards, 2006), size 116 cm (10th percentile). Length of arm stroke is 116 cm and height in sitting position of 63 cm, with a sitting size/standing size ratio corresponding to the average for his age. Facies with mild depressed nasal bridge, wide mouth, and thick lips. Complete mobility for the cervical spine. Apparent increase in lumbar lordosis. When flexing the trunk there is dorso-lumbar kyphosis (triangular), which is corrected in standing. In the upper extremities, there is a slight widening of wrists and interphalangeal joints, without any deformities in elbows or wrists. Joint mobility is normal and complete for hips, mild pain in the last degrees of abduction on the right hip. Both knees 12 degrees valgus and 30 mm of intermalleolar distance. Both feet with longitudinal arch within normality (Fig. 2).
The male sibling, an 8 year-old-boy, is an asymptomatic child, brought to examination because highlight similar clinical features as his sister.
He is the second pregnancy of same biparental family, regular full-term delivery, and no perinatal comments. Regular psychomotor development. Remarkable history of childhood recurrent upper respiratory tract infections, as result leading to adenoid hypertrophy diagnosis. During episodic pediatrician checkups, no growth alterations were detected. He has no other pathological history.
At orthopedic examination, weight 22.9 kg (10th percentile), height 119 cm (percentile 1) sitting height is 63 cm, relation sitting size/standing height, below percentile 3 [7]. Discrete facial findings like sister’s: Depressed nasal bridge, wide mouth, and thick lips; thorax with anterior prominence, no deviations in spinal axis. Knocking knees with 12 degrees valgus and 40 mm intermalleolar distance. Hindfoot with mild valgus and well-developed longitudinal arch (Fig. 3).
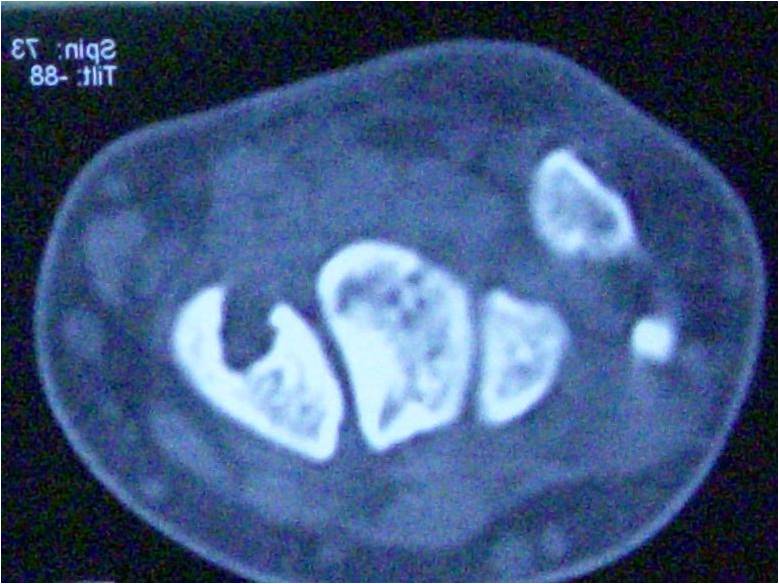
X-rays’ images for both sibs share mild dysostosis: Platyspondyly and kyphosis in thoraco-lumbar junction. Pelvic images show acetabular dysplasia and flattened, irregular femoral heads with fragmented epiphyses (Fig. 4). Radiographs of hands with triangular distal phalanges. All the above findings were compatible with spondylo-epiphyseal dysplasia.
As these findings on radiography do not fully correspond with Perthes and adding the history of respiratory tract infections, it lead us to perform an enzymatic study, which documented the deficiency of galactosamine-6-sulfate sulfatase, with homozygous variant c.199C> T p. (Arg67Trp) for the GALNs gene in both patients, confirming Morquio A disease in attenuated variant.
Cases shown warn us about wide and subtle variation in clinical presentation of MPS IV. The first patient in our report was previously approached with a suspect diagnosis of bilateral Perthes disease, based on pelvic radiographic findings. Antecedents on her brother were clue for suspicion. Presentation of a child who has “bilateral Perthes disease” as a reason for consultation, should alert the physician to expand differential diagnostic’s exercise. When skeletal changes that may coincide with localized bone diseases such as bilateral Perthes, there is a need to purposely seek changes in other joints [9]. Physicians involved in children management should consider lysosomal storage diseases as differential diagnoses. Diagnosis of specific skeletal dysplasia is a real challenge. Few doctors are used to treat large number of patients with these diseases to be considered experts in the field. Pelvic radiographic findings of diseases of very different origin may look alike. In clinical practice, it is very important to perform a complete physical examination in all children with skeletal alterations to suggest skeletal dysplasia. On physical examination, there are some signs that should alert us to the possibility such as short stature, angular misalignments of lower extremities, facial alterations, short neck, prominent thorax, deviations of the spine in the sagittal or coronal plane, and disproportion between extremities and trunk [10].
Perthes disease affects both hips only between 8 and 24% of patients. Wigg et al. report bilateral involvement in 55 of 425 children (13%) with Perthes. Of these two groups are distinguished: concurrent involvement (23 cases) and successive involvement (17 cases). Only 10 of the 23 concurrent cases had bilateral involvement at diagnosis in same radiographic stage [11]. The foregoing highlights the low possibility of bilateral Perthes with symmetric involvement in X-rays at diagnosis: only 2% of total Perthes and 18% of bilateral Perthes cases. In MPS IV as in most skeletal dysplasia’s, involvement of secondary ossification nucleus of the femoral head is usually symmetric [11, 12].
Changes in the femoral head seen in MPS are more like epiphyseal dysplasia’s than to Perthes disease: bone resorption is more lasting and typical Perthes’s stages of re-ossification and remodeling are not observed. In addition, necrosis remains in epiphysis whereas in Perthes disease it frequently affects epiphysis and metaphysis [9, 12, 13]. Our two siblings showed symmetric involvement of proximal femoral epiphyseal ossification center and consistent acetabular dysplasia, which warn us about not having a typical Perthes disease. Therefore, radiographic study was extended and multiple dysostoses in both patients were confirmed. Although little attention has been paid to acetabular changes, identification of these can be very useful in distinguishing between both conditions. Similar changes are observed in the acetabular side of the hip; however, acetabular dysplasia is usually more intense in MPS IV patients. Borowski et al., in a series of MPS IV patients between 8 and 16 years, reported an increase in the acetabular index on average 33 degrees, which is 12° higher than normal value for their age. In addition, computed tomography showed global acetabular deficiency, with predominant antero-superior dysplasia and no anteversion alterations [13]. Froberg et al. described that acetabular changes in Perthes disease usually few at beginning, but still develop through time. In same study, at diagnosis (average 6 or 7 years), acetabular index is not different from control group, but at end of follow-up (average 15 and 16 years), only patients classified in Stulberg III, IV, and V showed alterations in acetabular index: Average of 12° compared with 4° for control group [1]. In these two studies, a great difference can be identified comparing acetabular index of 33° in patients with MPS IV versus 12° for severe Perthes group. Our two cases reported had acetabular dysplasia in both hips (20 degrees for male and 25 in girl, who’s initial consultation was intermittent hip pain).
There are few reports of late diagnosed MPS IV patients (even in adulthood) initially treated as localized orthopedic problems. Fang-Kircher reported a 51-year-old male with hip osteoarthritis, initially diagnosed as Perthes disease at 13 years of age; hip articular replacement was planned when diagnosis of MPS IV was confirmed [10]. Prat describes a French woman, who from childhood was diagnosed with multiple skeletal alterations, and despite not having a definitive diagnosis, was operated throughout her life by alterations in the cervical spine, hips, knees and before confirming the enzymatic deficiency at age 38. In addition, they report her brother of 35 years with a similar phenotype [14]. Mendelsohn reports three cases of late diagnosed mucopolysaccharidosis, one MPS VI patient and one of the two MPS IV patients were initially diagnosed as bilateral Perthes disease. In all three cases, confirmation diagnosis was delayed because normal urinary glycosaminoglycans levels (uGAGs). Due to the lack of consistency to find high urinary levels of GAGs in the MPS, the authors suggest maintaining a high index of suspicion despite normal urinary levels of GAGs and underline the importance of molecular diagnosis [8, 15]. Rush reported a 15-year-old adolescent, with low back and right hip pain, initially diagnosed as avascular necrosis of the hip. He had a decrease in bone mineral density and vertebral changes initially considered as probable compression fractures. Later, platispondilia was confirmed. As in the previous report, uGAGs were normal. Diagnosis of MPS IV was made by sequencing exome due to the non-specificity mutation after he was approached as a probable spondyloepiphyseal dysplasia [7, 9].
More than 180 different mutations have been identified in the GALNS gene responsible to MPS IV. As an example, in genetic study for 163 MPS IV patients, Morrone, reported in 2014 a total of 39 “new” mutations not previously described [15]. Many mutations identified have been associated with attenuated variant of the disease. In 2007 Montaño recognize as many of 25% of their patients might be classified as attenuated phenotype. These patients are often diagnosed after 10 years of age; several years after the onset of symptoms (average 2.1 and 4.1 years of age) [3, 4]. Among the 326 participants on the International Registry of Morquio A in 2007, the diagnosis was made average at 4.7 years of age, while symptoms were initially recognized between 1 and 3 years (average of 2.1 years); In addition, 16% of cases were diagnosed between 5 and 10 years and 8% after 10 years (4). On the other hand, variability in phenotypic expression of the disease has been directly associated to residual enzymatic activity, especially in attenuated phenotype [2]. In addition to specific mutations, clinical factors have been associated with the classification of attenuated phenotype, particularly height and severity of symptoms; however, there are no unified criteria for categorization. Availability of clinical criteria for the early diagnosis of the disease could influence earlier identification in attenuated cases, which could help earlier treatment and prevent cumulative damage at older ages [6].
There are many factors that may delay early diagnosis in MPS IV. Children with disease appear healthy at birth. Subsequently, initial clinical manifestations vary due to great heterogeneity of mutations in GALNS gene, some of which have correlation between genotype-phenotype [6]. In Morquio’s disease, skeletal features are first clinical manifestations before diagnosis [4, 5]. Although phenotype is variable, multiple dysostosis is a constant finding on these patients. The X-ray of “bilateral Perthes disease at the same stage” should be an alert for a detailed patient study before planning any therapeutic intervention.
Clinical and radiographic assessment in children is not always easy. Minimal not characteristic changes of a disease should alert the physician to look for other causes of disease, which will improve patient’s prognosis at an early age.
References
- 1.Froberg L, Christensen F, Pedersen NW, Overgaard S. Radiographic changes in the hip joint in children suffering from Perthes disease. J Pediatr Orthop B 2012;21:220-5. [Google Scholar]
- 2.Montaño A, Sukegawa K, Kato Z, Carrozzo R, Di Natale P, Christensen E, et al. Effect of “attenuated” mutations in mucopolysaccharidosis IVA on molecular phenotypes of N-acetylgalactosamine-6-sulfate sulfatase. J Inherit Metab Dis 2007;30:758-67. [Google Scholar]
- 3.Leadley RM, Lang S, Misso K, Bekkering T, Ross J, Akiyama T, et al. A systematic review of the prevalence of Morquio a syndrome: Challenges for study reporting in rare diseases. Orphanet J Rare Dis 2014;9:173. [Google Scholar]
- 4.Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio a registry: Clinical manifestation and natural course of Morquio a disease. J Inherit Metab Dis 2007;30:165-74. [Google Scholar]
- 5.Colmenares-Bonilla D, Esquitin-Garduño N. Diagnosis of Morquio-a patients in Mexico: How far are we from prompt diagnosis? Intractable Rare Dis Res 2017;6:119-23. [Google Scholar]
- 6.Hendriksz CJ, Harmatz P, Beck M, Jones S, Wood T, Lachman R, et al. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab 2013;110:54-64. [Google Scholar]
- 7.Rush ET. Atypical presentation of mucopolysaccharidosis Type IVA. Mol Genet Metab Rep 2016;8:8-12. [Google Scholar]
- 8.Mendelsohn NJ, Wood T, Olson RA, Temme R, Hale S, Zhang H, et al. Spondyloepiphyseal dysplasias and bilateral Legg-Calve-Perthes disease: Diagnostic considerations for mucopolysaccharidoses. JIMD Rep 2013;11:125-32. [Google Scholar]
- 9.Andersen PE Jr., Schantz K, Bollerslev J, Justesen P. Bilateral femoral head dysplasia and osteochondritis. Multiple epiphyseal dysplasia tarda, spondylo-epiphyseal dysplasia tarda, and bilateral Legg-Perthes disease. Acta Radiol 1988;29:705-9. [Google Scholar]
- 10.Fang-Kircher SG, Böck A, Fertschak W, Schwägerl W, Paschke E. Morquio disease in a patient diagnosed as having Perthes disease for 38 years. J Inherit Metab Dis 1995;18:94-5. [Google Scholar]
- 11.Wiig O, Huhnstock S, Terjesen T, Pripp A, Svenningsen S. The outcome and prognostic factors in children with bilateral Perthes’ disease: A prospective study of 40 children with follow-up over five years. Bone Joint J 2016;98-B:569-75. [Google Scholar]
- 12.Ikegawa S, Nagano A, Nakamura K. A case of multiple epiphyseal dysplasia complicated by unilateral Perthes’ disease. Acta Orthop Scand 1991;62:606-8. [Google Scholar]
- 13.Borowski A, Thacker MM, Mackenzie WG, Littleton AG, Grissom L. The use of computed tomography to assess acetabular morphology in Morquio-Brailsford syndrome. J Pediatr Orthop 2007;27:893-7. [Google Scholar]
- 14.Prat C, Lemaire O, Bret J, Zabraniecki L, Fournie B. Morquio syndrome: Diagnosis in an adult. Joint Bone Spine 2008;75:495-8. [Google Scholar]
- 15.Morrone A, Tylee KL, Al-Sayed M, Brusius-Facchin AC, Caciotti A, Church HJ, et al. Molecular testing of 163 patients with Morquio A (Mucopolysaccharidosis IVA) identifies 39 novel GALNS mutations. Mol Genet Metab 2014;112:160-70. [Google Scholar]