
With increased expression of SCN11A, patients with CIP may experience heterotopic ossification at a higher rate when compared to other patients undergoing similar procedures and this may help link an association between HO, CIP, Substance P and the DRG for further research.
Dr. Gabriel Makar, Department of Pediatric Orthopaedics, PGY-4 Orthopaedic Surgery Resident Geisinger Health System, Danville, Pennsylvania 17821, United States. E-mail: gmakar1@som.geisinger.edu
Abstract
Introduction: A male child with congenital insensitivity to pain (CIP) due to a novel de novo L369P mutation in the SCN11A gene was found to have significant bilateral hip flexion contractures, followed by severe heterotopic ossification after contraction release. This is the first report to describe a patient with this specific mutation and subsequent clinical course.
Case Report: A male child with CIP due to de novo L369P mutation in the SCN11A gene was found to have significant bilateral hip flexion contractures. The patient underwent bilateral hip contracture releases to improve his standing ability after failure of conservative treatment. In the coming months he developed significant heterotopic ossification that eventually bridged from the left pelvis to the left femur.
Conclusion: Heterotrophic ossification (HO) in patients with CIP is a rare and poorly understood clinical manifestation. Our report describes a rare and aggressive manifestation of HO in a patient with CIP.
Keywords: Congenital insensitivity to pain, heterotrophic ossification, pediatrics.
Congenital insensitivity to pain (CIP) is part of a group of particularly rare hereditary sensory and autonomic neuropathies (HSAN). CIP is reported to have an incidence of 1 in every 125 million newborns [1]. CIP is typically inherited in an autosomal recessive manner due to mutations genes that code for voltage-gated sodium channels (SCN9A, SCN11A, PRDM12) or a defect in tyrosine kinase-1 receptor (NTK-1 or NGF) [2]. However, several cases of this condition have displayed an autosomal dominant pattern, or spontaneous mutations without any indication of hereditability [10]. Through these defects, development of portions of the afferent sensory system responsible for relying nociceptive stimuli is compromised. In individuals with mutations to NTRK1, anhidrosis is often associated with CIP due to loss of autonomic sympathetic neurons that innervate eccrine sweat glands [3]. Patients with this condition typically are able to perceive all sensations besides pain. Subsequently, patients affected by this condition have diminished sensation to pain and are suspectable to fractures, deep skin infection, osteomyelitis, and joint deformation, mostly of the lower extremities [1,4]. The orthopedic manifestations of this condition occur from the self-mutilation behavior that occurs due to loss of pain sensation that typically prevents damage and defected bone metabolism. The loss of nociceptive fibers in the skeletal system negatively affects bone metabolism, leading to an increased risk for bone fractures [5,6]. The management of this CIP is not clear cut, with treatment mostly focused on addressing symptomatology [7]. In this case, we present a patient with a rare mutation causing CIP that developed progressive bilateral hip heterotrophic ossification (HO) soon after bilateral hip flexor release.
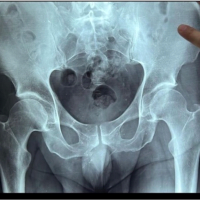
A 9-month-old boy first presented for a neurodevelopmental pediatric evaluation due to significant self-biting of the tongue, lower lip, and fingertips. Patient first began to develop signs of self-harm approximately at 5 months when abrasions to the tongue were noted by the pediatric dentist. The child was born to a gravida 3, para 1 mother; there was one prior miscarriage. Fetal movement was normal through the pregnancy, and the baby was born by spontaneous vaginal delivery at 39 weeks. He did not have any other major neonatal complications. During this visit, the patient was found to have notable global developmental delays and had significant hypotonia of the lower extremities. Patient was able to roll over from prone to supine position at 6 months. The patient also had delays in language and fine motor development and was unresponsive to painful stimuli (lack of response to blood obtained from toe). Lastly, the patient exhibited poor weight gain due to poor oral feeding. Laboratory work at this visit eliminated the possibility of Lesch-Nyhan syndrome and magnetic resonance imaging (MRI) of the brain was unremarkable. 2nd neurodevelopmental evaluation at 12 months old, revealed the patient continued to exhibit global developmental delays and neuromotor abnormalities (hypotonia and hyporeflexia). His self-biting progressed to causing visible tissue injury to his fingertips and his poor feeding persisted. Whole exome sequencing was performed after this visit and revealed a mutation in the SCN11 gene. At 16 months of age the patient began nasogastric feedings due to poor weight gain in frequent spitting up of food. A G-tube was placed in later weeks due to chronic malnutrition and inability to tolerate oral feedings. Patient’s first orthopaedic visit occurred at 17 months of age and revealed he can now sit independently but cannot pull to stand or walk yet. His physical exam revealed full range of motion of the shoulders, elbows, wrists and hands with normal alignment and stability. Muscle tone and reflexes were intact in the upper extremities. His lower extremities show equal leg lengths and symmetric abduction to a peroneal angle of 150°. Hip rotation shows internal rotation to 30° and external rotation to 50°. There was diffuse hypotonia in both lower extremities. A discussion was had at the time to defer bracing until the child can walk to prevent any impedance to his motor development. At the age of 25 months, the patient returned with normal mobility, except at the hip where he showed increased flexion contracture. At approximately 4 years of age, the patient was able to walk 10 steps independently with a 50° flexion contracture at both hips. Follow up at 4.5 years old revealed little improvement in hip flexion contracture despite persistent stretching by the patient’s physical therapist and mother since the patient was 25 months old. Computed tomography of the hip at 4.5 years old demonstrated well-formed acetabulum anteriorly with minor deficits posteriorly, confirming a soft issue contracture (Fig. 1). Operative treatment to release contracture of both hips was discussed with family since nonoperative management did not prevent progression of the patient’s contractures. Patient underwent surgery at approximately 4 years and 9 months of age. Intraoperatively, a hip arthrogram was first performed to assess the overall contour of the hip, femoral head, and acetabulum anatomy. During soft tissue dissection the sartorius, tensor fascia lata and rectus femoris were found to be excessively tight. Bilateral sartorius and rectus femoris tenotomies were performed in addition to the recession of bilateral iliopsoas muscles. Partial release of bilateral tensor fascia lata was also performed with anterior hip capsule release and reconstruction of both hips to allow the hips to achieve a neutral position. He was maintained for 6 weeks in short leg casts connected with a bar to lock the hips in internal rotation to reduce risk of anterior dislocation of hips. Approximately 2 months post-op he was able to stand longer and straighter than prior to surgery. Radiographs of the hips at this visit revealed signs of HO around the right femur, left acetabulum, and left femur. However, over the course of months he developed a diminishing walking ability. Repeat imagining approximately 6 months post-operatively demonstrated heterotopic ossification around the right femur, left acetabulum and left femur that later bridged from the left pelvis to femur (Fig. 2-4). The patients last presented 14 months post-op with acutely elevated inflammatory markers (erythrocyte sedimentation rate [43 mm/h] and C-reactive protein [8 mg/L]) as well as a left hip swelling. An MRI revealed an effusion in his left hip. At this visit his range of motion of the right hip was flexion from 60° to 80°. Left hip was fused at 70° of flexion. An aspiration of his left hip was later negative for infection. The aspiration was complicated by the overly heterotopic ossification as well as new periosteal bone formation appreciated intra-operatively using fluoroscopic imaging.
Partial release of bilateral tensor fascia lata was also performed with anterior hip capsule release and reconstruction of both hips to allow the hips to achieve a neutral position. He was maintained for 6 weeks in short leg casts connected with a bar to lock the hips in internal rotation to reduce risk of anterior dislocation of hips. Approximately 2 months post-op he was able to stand longer and straighter than prior to surgery. Radiographs of the hips at this visit revealed signs of HO around the right femur, left acetabulum, and left femur. However, over the course of months he developed a diminishing walking ability. Repeat imagining approximately 6 months post-operatively demonstrated heterotopic ossification around the right femur, left acetabulum and left femur that later bridged from the left pelvis to femur (Fig. 2-4). The patients last presented 14 months post-op with acutely elevated inflammatory markers (erythrocyte sedimentation rate [43 mm/h] and C-reactive protein [8 mg/L]) as well as a left hip swelling. An MRI revealed an effusion in his left hip. At this visit his range of motion of the right hip was flexion from 60° to 80°. Left hip was fused at 70° of flexion. An aspiration of his left hip was later negative for infection. The aspiration was complicated by the overly heterotopic ossification as well as new periosteal bone formation appreciated intra-operatively using fluoroscopic imaging. 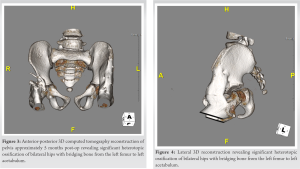 Patient consent was obtained prior to production of this case report.
Patient consent was obtained prior to production of this case report.
This case presents a male child with a de novo L369P mutation of the alpha subunit of the sodium voltage gated channel (SCN11A), indicating a hereditary sensory and autonomic neuropathy type 7 (HSAN7). Months after a surgical procedure to treat his persistent hip contracture, the patient developed relatively rapid and diffuse heterotopic ossification of the bilateral hip joints that compromised the increased range of motion achieved from the surgery. To the authors’ best knowledge, this is one of the first case reports to describe a patient with CIP who subsequently developed a rather severe HO after a surgical procedure. CIP is an uncommon condition that typically leads to loss of pain sensation, which is more pronounced in the lower extremities. This condition was first discussed by Dearborn in 1932 where he described a 44-year-old man who did not recall any sensation of pain besides headache throughout his life [9]. In recent years, some of the specific mutations that lead to the manifestation of this condition have been elicited. It is now known CIP occurs secondary to nonfunctional nociceptors or failure of the nociceptors to develop [10]. Due to the absence of perception of pain, patients with this condition are vulnerable to many conditions, beginning at birth. The common orthopedic manifestations of CIP include progressive joint swelling, painless fractures, Charcot arthropathy, joint dislocations, and osteomyelitis secondary to loss of protective somatic pain sensation [8]. These orthopedic manifestations occur traditionally later in the course of the illness as wear and tear damage of bone and associated structures take time to develop. A study by Schon et al. recommends at least a once-a-year bone health assessment for patients with CIP to prevent and treat complications [10]. However, the association between HO and CIP in the current literature is relatively weak. Heterotopic ossification is the development of extra skeletal bone secondary to trauma, neurologic injury, and excessive intracellular signaling [11]. The development of HO is thought to center around the function of the dorsal root ganglion (DRG) after trauma. Following a traumatic incident DRG releases the neuro-inflammatory cytokine Substance P, which recruits stem cells to initiate the formation of HO [11]. The DRG has high expression of SCN11A, providing a link between HO and CIP. The ramifications of this defected channel in the DRG have shown to increase the firing rate of neuronal cells in the DRG, rendering hyperactive DRG neurons that may release an increased amount of substance P [12,13]. However, there has been no published studies to further confirm this speculative assessment. More studies are required to further elucidate the pathogenies of HO in patients with CIP.
Our study describes a patient with HSAN7 who underwent multiple operations for their hip flexion contractures found later to have extensive heterotopic ossification. Our research finds an association between heterotopic ossification and CIP through a high expression of SCN11A. With increased expression of SCN11A, patients with CIP may experience heterotopic ossification at a higher rate when compared to other patients undergoing similar procedures. Further research and larger patient cohorts are necessary to better understand the association between CIP and their increased risk for HO.
Patients with rare diseases or disorders may have unusual complications secondary to the complex nature of these patients. Heterotopic ossification is a rare complication in patients with CIP and the SCN11A mutation may help provide an avenue for researchers to better understand the association with Substance P, pain sensation and heterotopic ossification.
References
- 1.Kurnaz R, Asci M, Balta O, Aytekin K, Gunes T. Congenital insensitivity to pain syndrome accompanied by neglected orthopedic traumas and complications. Arch Clin Cases 2017;4:27-34. [Google Scholar | PubMed]
- 2.Spiteri M, Mifsud M, Azzopardi T, Giele H. Population study of hand and wrist manifestations of congenital insensitivity to pain. Hand 2020;17:155–161. [Google Scholar | PubMed]
- 3.Bar-On E, Weigl D, Parvari R, Katz K, Weitz R, Steinberg T. Congenital insensitivity to pain. Orthopaedic manifestations. J Bone Joint Surg Br 2002;84:252-7. [Google Scholar | PubMed]
- 4.Derwin KA, Glover RA, Wojtys EM. Nociceptive role of substance-P in the knee joint of a patient with congenital insensitivity to pain. J Pediatr Orthop 1994;14:258-62. [Google Scholar | PubMed]
- 5.Hill EL, Elde R. Distribution of CGRP-, VIP-, DβH-, SP-, and NPY-immunoreactive nerves in the periosteum of the rat. Cell Tissue Res 1991;264:469-80. [Google Scholar | PubMed]
- 6.Pérez-López LM, Cabrera-González M, Gutiérrez-de la Iglesia D, Ricart S, Knörr-Giménez G. Update review and clinical presentation in congenital insensitivity to pain and anhidrosis. Case Rep Pediatr 2015;2015:589852. [Google Scholar | PubMed]
- 7.Schwarzkopf R, Pinsk V, Weisel Y, Atar D, Gorzak Y. Clinical and genetic aspects of congenital insensitivity to pain with anhidrosis. Harefuah 2005;144:433-7, 453, 452. [Google Scholar | PubMed]
- 8.Bronfen C, Bensahel H, Teule JG. Orthopedic aspects of congenital insensitivity to pain. Chir Pediatr 1985;26:193-6. [Google Scholar | PubMed]
- 9.Dearborn GV. A case of congenital general pure analgesia. J Nerv Ment Dis 1932;75:612-5. [Google Scholar | PubMed]
- 10.Schon KR, Parker AP, Woods CG. Congenital insensitivity to pain overview. In: GeneReviews®. Seattle, WA: University of Washington; 2020. [Google Scholar | PubMed]
- 11.Kan L, Lounev VY, Pignolo RJ, Duan L, Liu Y, Stock SR, et al. Substance P signaling mediates BMP‐dependent heterotopic ossification. J Cell Biochem 2011;112:2759-72. [Google Scholar | PubMed]
- 12.Zhang XY, Wen J, Yang W, Wang C, Gao L, Zheng LH, et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet 2013;93:957-66. [Google Scholar | PubMed]
- 13.King MK, Leipold E, Goehringer JM, Kurth I, Challman TD. Pain insensitivity: Distal S6-segment mutations in NaV1.9 emerge as critical hotspot. Neurogenetics 2017;18:179-81. [Google Scholar | PubMed]



Related Articles in Journal of Orthopaedic Case Reports
 August 6, 2024 Intra-articular Osteochondroma Of The Hip. Total Hip Replacement After Tumor Resection In A Young Adult – A Case Report
August 6, 2024 Intra-articular Osteochondroma Of The Hip. Total Hip Replacement After Tumor Resection In A Young Adult – A Case Report October 9, 2012 Editorial: Personalised Journal, Personalised Network and Clinical Decision Making (CDM)
October 9, 2012 Editorial: Personalised Journal, Personalised Network and Clinical Decision Making (CDM) July 10, 2020 Management of Proximal Thoracic Kyphoscoliosis with Early Myelopathy in a Young Adult with Neurofibromatosis Type 1: A Case Report and Review of Literature
July 10, 2020 Management of Proximal Thoracic Kyphoscoliosis with Early Myelopathy in a Young Adult with Neurofibromatosis Type 1: A Case Report and Review of Literature June 1, 2026 Staged Reconstruction for Dorsal Hand and Wrist Shear Injury: A Case Report
June 1, 2026 Staged Reconstruction for Dorsal Hand and Wrist Shear Injury: A Case Report