
Freeman-Sheldon Syndrome is a rare congenital anomaly that should be differentiated from another syndrome of the close resemblance, Sheldon Hall syndrome, and Schwartz Jampel syndrome.
Dr. Santanu Kar, Department of Orthopedics, All India Institute of Medical Sciences, New Delhi, India. E-mail: karsantanu109@gmail.com
Abstract
Introduction: Freeman-Sheldon syndrome (FSS), also known as the distal arthrogryposis (DA) type 2A, is a rare congenital anomaly. We report a unique case of the DA type 2A with mixed clinical features and the unusual presentation of bilateral congenital dislocation of the knee but had unassisted stiff knee gait.
Case Report: A 5-year-old female child presented to the clinic with the complaint of inability to bend both knees since birth. She had an unassisted bipedal gait, but could not squat, cross-leg sit, run, and climb stairs without assistance. Her youngest brother had a similar presentation but succumbed to death at the age of 5 months due to respiratory distress. Clinical features were in the favor of FSS. Her serum creatinine kinase level was normal and the electromyography of bilateral tibialis anterior and abductor pollicis brevis was not suggestive of the myotonia. Radiograph of the skull showed cooper beaten skull appearance whereas bilateral pelvis with the hip showed following changes in the right hip; decrease femoral epiphysis height, horizontal proximal femoral physis, and the coxa brevia. She was initially managed conservatively by weekly stretching, manipulation, and casting. As a result, she could flex her knee up to 20°. Although the quadricepsplasty might be helpful for the persistent extension deformity, there was marked quadriceps weakness which could make it harder for the child to stand and walk. In addition, the abnormal muscle physiology in FSS may result in unfavorable outcomes after the surgery. Moreover, a consideration of the surgical aspect is not free of risks which include difficult endotracheal intubation, vein access, and malignant hyperthermia.
Conclusion: FSS is a rare congenital anomaly that should be differentiated from another syndrome of the close resemblance, Sheldon Hall syndrome and Schwartz Jampel syndrome which are other rare autosomal recessive disorders characterized by myotonia and the chondrodysplasia. Conservative management has still a role in bilateral knee involvement especially if the patient is an independent walker.
Keywords: Freeman-Sheldon syndrome, arthrogryposis, quadricepsplasty, congenital contractures, gait.
Freeman-sheldon syndrome (FSS), named after Freeman, an orthopedic surgeon, and sheldon, a pediatrician. FSS was described as the syndrome of cranio-carpo-tarsal dystrophy in 1938. Also known as the distal arthrogryposis (DA) type 2A, the syndrome is rarely characterized by multiple congenital contractures (MCCs), that is, restricted movement around two or more body areas at birth, abnormalities of the craniofacial area, skeletal malformations, and defects of the hands and feet [1]. The occurrence of FSS is usually sporadic, but autosomal dominant and autosomal recessive transmissions have also been reported [2].
The hallmark clinical features of FSS include microstomia, whistling-face appearance (pursed lips), H-shaped chin dimpling, nasolabial folds, and two or more contractures of hands and feet [3]. Infants with Freeman-Sheldon syndrome often have skeletal abnormalities including progressive kyphoscoliosis, contractures of the knees, shoulders, and/or hips resulting in limited movement of respective joints or dislocation [1]. The stiff knee gait is characterized by the diminished knee peak flexion during the swing phase. The index case has a typical presentation of a probable non-sporadic form of FSS with dislocation of bilateral knee joints and an unassisted stiff knee gait. Parents were concerned about the limitation of the ambulatory status. Hence, surgical intervention was deferred because of likely loss of standing/walking.
A 5-year-old female child, born out of the non-consanguineous marriage of young healthy parents; presented to the orthopedic outpatient clinic with complaints of inability to bend both the knees since birth which altered squatting and cross leg sitting. Although she had an unassisted bipedal stiff knee gait, she could not run and climb stairs without assistance. The child had normal developmental milestones and fair scholastic performance. There was also a defect in speech which caused mumbling voice, difficulty in social interaction, maintenance of dental hygiene, and feeding. However, she was able to wear her clothes herself, brushing her teeth, transferring, and eating but need assistance with taking bath and with toilet activities.
Antenatal history was insignificant. She was born out of full-term vaginal breech delivery with a birth weight of 2.2 kg. The child was later diagnosed to have bilateral CTEV and genu recurvatum, for which weekly stretching, manipulation, and casting were done for around 2 years. She had two younger brothers; the youngest had a similar clinical presentation at birth and died at the age of 5 months due to respiratory distress. The mother had a spontaneous abortion during her fourth pregnancy at 4 months of gestation. There was no history of similar complaints or primary neurological/muscular disorders in the family.
On examination, the patient had a bodyweight of 10.4 kg, a height of 98 cm, and a head circumference of 42 cm. She had short stature, mask-like face, bilateral ptosis of eyes, microstomia, pursed lips, high arched palate, absent skin creases at the bilateral elbow, knee, and wrist joints, clasped thumb deformity, that is, inability to voluntarily extend and abduct bilateral thumbs. The patient could flex her knee up to 20°. She had pectus excavatum, scoliosis of the thoracolumbar spine with convexity towards the left side, and increased right trunk – arm distance. She also had bilateral broad and wide heels as shown in (Fig. 1, 2, 3). Her ophthalmological examination had revealed poor power of bilateral levator palpebrae superioris muscle, hypermetropia with +2.50 D spherical correction in the right eye and +3.00 D in the left eye. Other features associated with the syndrome such as webbed neck, H-V dimpled chin, mal-occluded teeth, and hearing loss were absent.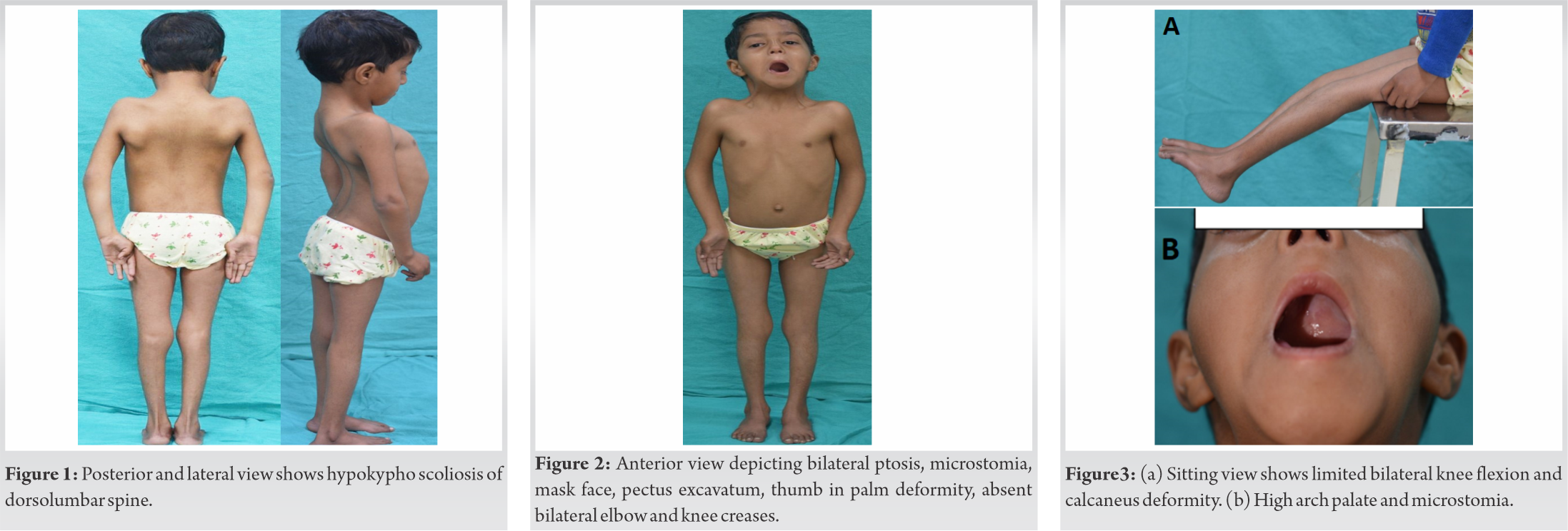
Routine hematological investigations including serum creatine phosphokinase levels were normal. Electromyography studies of the bilateral tibialis anterior and the abductor pollicis brevis reported a normal insertional activity, no positive sharp waves, fibrillation, and myotonic discharge. The lateral radiographic view of the skull showed a copper-beaten appearance as shown in (Fig. 4), which was most likely because of the craniosynostosis secondary to microcephaly. Her pelvis with bilateral hips showed the following changes in the right hip-decreased femoral epiphysis height, horizontal proximal femoral physis, and coxa breva as shown in (Fig. 5). Her lateral radiographic view of the bilateral knee showed genu recurvatum as shown in (Fig. 6). A skeletal survey showed the delayed appearance of the ossific nucleus of the proximal fibular epiphysis, medial epicondyle of humerus and patella. However, radiographs of the thoracolumbar spine showed no wedging or hemivertebrae or any other structural scoliotic changes.
The child has been under observation for the past 5 years and the past follow-up was 9 months ago in our department. The child has been stable and no significant worsening has occurred over the past 5 years. Since the clinical presentation of FSS may vary in each individual, we cannot define a particular algorithm of treatment. For our patient, we managed the child initially with manipulation and casting till the age of 2 years, followed by walking with knee stabilizing brace and passive stretching exercises. The child is currently able to do most of the activity of daily living. Due to the unexpected COVID-19 pandemic situation, the child could not be followed up after the last follow-up in March, 2019.
Due to the extreme rarity of this syndrome, and as this is the only case reported in literature from the region, and our center, it may not be possible to define a particular algorithm. Rather we had suggested that the management should be modified on a case to case basis, as highlighted in this case report.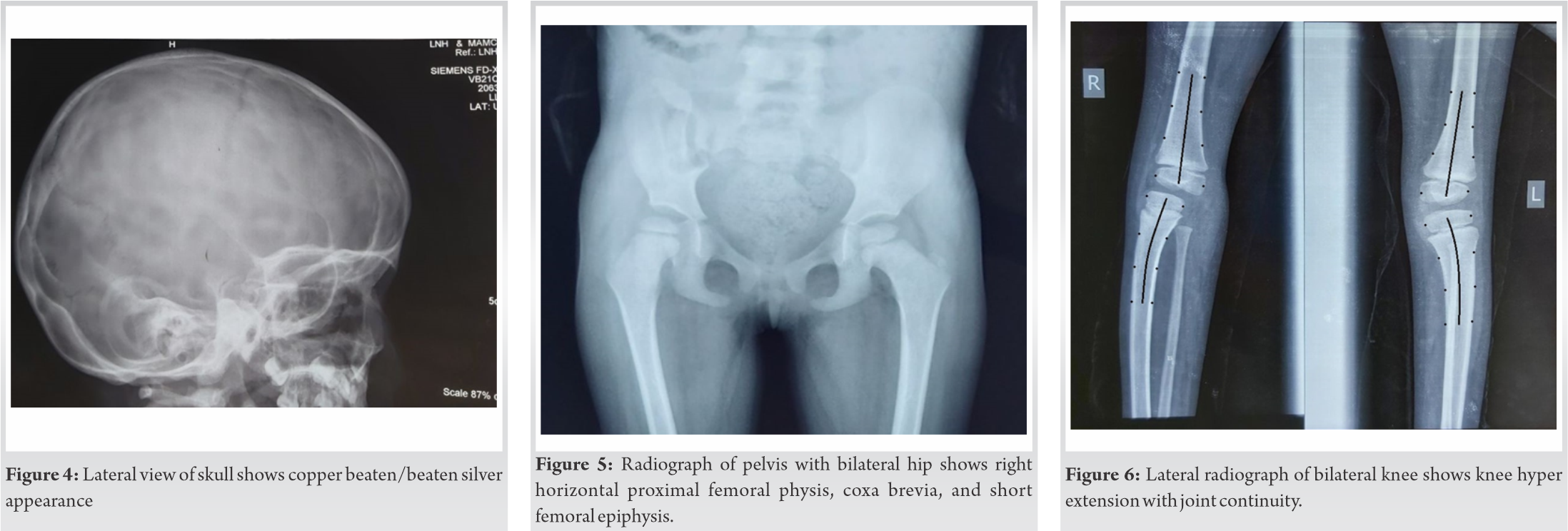
Congenital contractures in children are broadly divided into two groups: Isolated congenital contractures (e.g., clubfoot) and MCCs. Approximately 1 in 3000 children are born with MCCs [4]. MCCs which present congenitally, non-progressive, and involve more than one body area/joints defined as arthrogryposis. At least 150 specific entities have been recognized that have MCCs and are classified under arthrogryposis. Their best classification includes – amyoplasia, DA, and disorders with limb involvement and the central nervous system dysfunction [5].
Hall et al. [6] classified DA into ten hierarchically related disorders (i.e., DA1–DA10) according to the proportion of features they shared (i.e., DA1 is more similar to DA2 than it is to DA3, and so on). Features shared among all DA that helps in increasing the accuracy of diagnoses and improving patient management, including a consistent pattern of hand and foot involvement, limited involvement of proximal joints, and variable expressibility. Freeman-sheldon syndrome or whistling face-windmill vane hand syndrome is described by clinical diagnostic criteria devised by Stevenson et al. in 2006, which also helps in differentiating it from a closely resembling entity Sheldon-Hall syndrome (SHS). He mentioned the clinical criteria for FSS diagnosis: microstomia, whistling-face appearance (pursed lips), H- or V-shaped chin dimpling, prominent nasolabial folds, and major contractures in two or more body regions [3]. In addition, spinal deformities, metabolic, gastroenterological problems, other dysmorphic craniofacial characteristics, visual, and auditory impairments are frequent findings.
The index case was diagnosed based on clinico-radiological examination. Various clinical features in the index case that helped to make a diagnosis of FSS instead of SHS are summarized in (Table 1) [7]. Various etiological factors have been postulated for the occurrence of the syndrome; breech vaginal delivery was one risk factor that was present apart from the hereditary cause, which could be explained by the occurrence of similar complaints in the youngest sibling. Preferable involvement of females is noted in the majority of case reports [8].
The etiology of congenital knee dislocation includes short quadriceps tendon, tight anterior knee capsule hypoplastic suprapatellar pouch with anterior subluxation of hamstring tendons [9]. Isolated congenital dislocation of the knee (CDK) can be reduced generally by non-surgical means through serial casting or Pavlik harness. In contrast, CDK with AMC or Larsen syndrome after manipulation and casting shows residual subluxation which can result in a stiff knee gait. A fixed extended knee allows stable standing but makes sitting difficult. If the knee is stiff, a position of about 15° of flexion is the best for both standing and sitting. The management of the syndrome should aim toward maintaining/improving function and independence. In 5–23% of knee corrective surgeries required which includes quadricepsplasty, femoral shortening osteotomy. Several techniques have been reported for quadricepsplasty; percutaneous recession, and mini-open to formal open lengthening. Several authors have reported knee instability following surgery for CDK with a frequency as high as 78% [10]. Due to the abnormal muscle physiology in FSS, therapeutic measures may have unfavorable outcomes [11].
Even the considerable surgical interventions (for contractures, microstomia) are of great concern due to well-known anesthetic complications. People with FSS have an increased risk of developing a syndrome of high-grade fever, tachycardia, muscle rigidity, rhabdomyolysis, and metabolic acidosis, known as malignant hyperthermia in response to certain drugs such as muscle relaxants and anesthetic gases used during surgery [12]. Furthermore, structural anomalies of the oropharynx and upper airways, which are commonly diagnosed in these patients, are a constant concern in cases requiring general anesthesia. Difficult endotracheal intubation and venous access complicate operative decisions in many patients of FSS [13].
After knowing all these facts, we treated our patient with conservative modalities which included stretching followed by casting, splinting, paddling, muscle strengthening exercises, and regular follow-ups. The child was not operated on as it would have been challenging and could have led to the recurrence of deformity and loss of ambulation. The prognosis was fair as she was able to walk without support and ankle joints were spared. However, she is likely to have avascular necrosis of the femoral head, chondrolysis, osteochondritis dissecans, secondary arthritis, dislocation of the hip, trendelenburg gait, and limb length discrepancy. Regular follow-up is needed keeping the probable complications in mind with frequent radiographs. Osteotomies and soft tissue releases can be performed if necessary, to prevent such complications.
Individuals with FSS should be counseled that the recurrence risk for FSS is 50%, whereas the parents of a child with FSS have a recurrence risk only slightly higher than the population risk. Prenatal diagnosis can be frequently established via ultrasound examination, although contractures in DA disorders are frequently not evident until 18–24 weeks of gestation. In addition, because of the phenotypic similarity among DA1, SHS, and FSS, distinguishing among them in the prenatal period is challenging. Identification of the genetic basis of FSS will facilitate the prenatal diagnosis by making direct testing of DNA from a sample of chorionic villus or amniotic fluid possible [3]. Preconception testing of polar bodies and prenatal diagnosis can be achieved by targeted mutation testing of the myosin heavy chain 3 gene (MYH3; MIM #160720). About 7% of clinical FSS phenotypes are unaccounted for by currently documented allelic variations [14] and some MYH3-associated allelic variations do not always produce an FSS phenotype [15]. We could not perform genetic testing in our case due to the unavailability of resources. The differential diagnosis of FSS is tabulated in Table 1.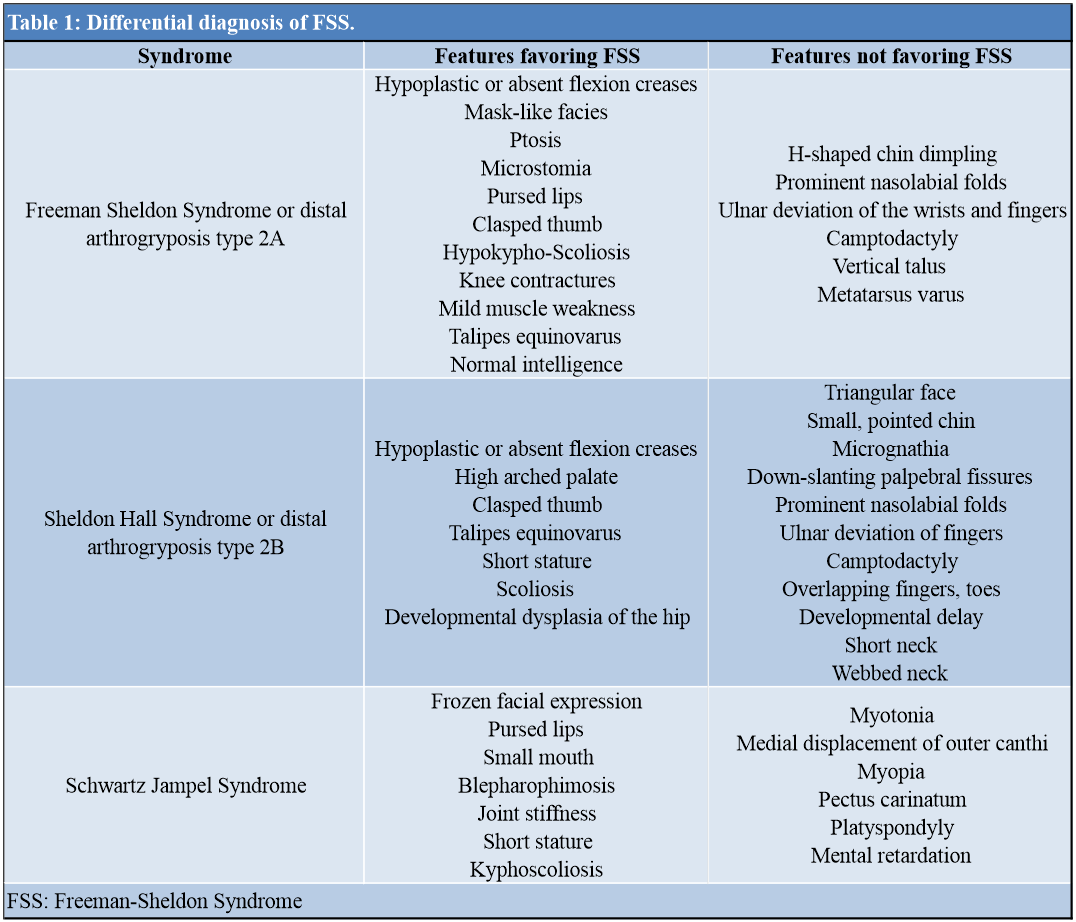
We concluded that FSS better be diagnosed on the clinicoradiological background. It should be differentiated from SHS or Schwartz Jampel syndrome which has implications both on prognosis and treatment plan.
The distinction of overlapping clinical features with myotonic and other hereditary diseases. Non-operative intervention still has a role in the management of the special type of arthrogryposis like FSS.
References
- 1.Available from: https://rarediseases.org/rare-diseases/freeman-sheldon-syndrome [Accessed on 12th September, 2021] [Google Scholar | PubMed]
- 2.Herring JA. Tachijain’s Pediatric Orthopaedics. 5th ed. Amsterdam, Netherlands: Elsevier; 2014. [Google Scholar | PubMed]
- 3.Stevenson DA, Carey JC, Palumbos J, Rutherford A, Dolcourt J, Bamshad MJ. Clinical characteristics and natural history of Freeman-Sheldon Syndrome. Pediatrics 2006;117:754-62. [Google Scholar | PubMed]
- 4.Fahy MJ, Hall JG. A retrospective study of pregnancy complications among 828 cases of arthrogryposis. Genet Couns 1990;1:3-11. [Google Scholar | PubMed]
- 5.Staheli LT, Hall JG, Paholke DO. Arthrogryposis a Text Atlas. 1st ed. Cambridge, United Kingdom: Cambridge University Press; 1998. [Google Scholar | PubMed]
- 6.Hall JG, Reed SC, Greene G. The distal arthrogryposes: Delineation of new entities: Review and nosologic discussion. Am J Med Genet 1982;11:185-239. [Google Scholar | PubMed]
- 7.Polling MI, Corado JA, Chamberlain RL. Findings, phenotypes, and outcomes in Freeman-Sheldon and Sheldon-Hall syndromes and distal arthrogryposis types 1 and 3: Protocol for systematic review and patient-level data meta-analysis. Syst Rev 2017;6:46. [Google Scholar | PubMed]
- 8.Laishley RS, Roy WL. Freeman-Sheldon syndrome: Report of three cases and the anesthetic implications. Can Anaesth Soc J 1986;33:388-93. [Google Scholar | PubMed]
- 9.Ooishi T, Sugioka Y, Matsumoto S, Fuji T. Congenital dislocation of knee: Its pathological features and treatment. Clin Orthop Relat Res 1993;287:187-92. [Google Scholar | PubMed]
- 10.Oetegn ME, Walick KS, Tulchin K, Karol LA, Johnston CE. Functional results after surgical treatment for congenital knee dislocation. J Pediatr Orthop 2010;30:216-23. [Google Scholar | PubMed]
- 11.Jones R, Dolcourt JL. Muscle rigidity following halothane anesthesia in two patients with Freeman-Sheldon syndrome. Anesthesiology 1992;77:599-600. [Google Scholar | PubMed]
- 12.Sobrado CG, Ribera M, Marti M, Erdocia J, Rodriguez R. Freeman-Sheldon syndrome: Generalized muscular rigidity after anesthetic induction. Rev Esp Anestesiol Reanim 1994;41:182-4. [Google Scholar | PubMed]
- 13.Gurjar V, Parushetti A, Gurjar M. Freeman Sheldon syndrome presenting with microstomia: A case report and literature review. J Maxillofac Oral Surg 2013;12:395-9. [Google Scholar | PubMed]
- 14.Toydemir RM, Rutherford A, Whitby FG. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat Genet 2006;38:561-5. [Google Scholar | PubMed]
- 15.Chamberlain RL, Poling MI, Portillo AL, Morales A, Ramirez RR, McCormick RJ. Freeman Sheldon syndrome in a 29 year-old woman presenting with rare and previously undescribed features. Case Reports 2015;2015:bcr2015212607. [Google Scholar | PubMed]
Related Articles in Journal of Orthopaedic Case Reports
 December 6, 2020 A Missed Case of Osteoid Osteoma of the Acetabulum Treated with a Novel Computed Tomography-Guided Technique – A Case Report
December 6, 2020 A Missed Case of Osteoid Osteoma of the Acetabulum Treated with a Novel Computed Tomography-Guided Technique – A Case Report February 1, 2026 Humerus Non-union with Segmental Bone Loss Treated with 3D-printed Prosthesis: A Case Report
February 1, 2026 Humerus Non-union with Segmental Bone Loss Treated with 3D-printed Prosthesis: A Case Report November 1, 2025 The Indian Joint Registry: Building Trust, Safety, and Excellence in Indian Orthopedics
November 1, 2025 The Indian Joint Registry: Building Trust, Safety, and Excellence in Indian Orthopedics January 28, 2015 Successful Correction of Idiopathic Bilateral Flexion Deformity of Knee: A Rare Case Report
January 28, 2015 Successful Correction of Idiopathic Bilateral Flexion Deformity of Knee: A Rare Case Report