
Metastatic neuroblastoma should be considered one of the differentials of lytic lesions of extremities in children.
Dr. Mukesh Kumar, Department of Radiodiagnosis, All India Institute of Medical Sciences Bhopal, Bhopal, Madhya Pradesh, India. E-mail: mukesh79655@gmail.com
Introduction: Neuroblastoma is an embryonic tumor of the peripheral sympathetic nervous system. It is the most common extracranial solid tumor of childhood and accounts for up to 15% of all pediatric cancer fatalities. The manifestation of neuroblastoma is variable depending on the location of the tumor and the presence or absence of paraneoplastic syndromes. Neuroblastoma has a great propensity to metastasize to multiple organs. The most common site of metastasis is the bone and bone marrow. Ewing sarcoma is a rare small round blue cell tumor originating from neuroectoderm. It is the most common primary bone tumor of adolescents. The most common anatomical sites of involvement include the pelvis, axial skeleton, and femur; however, Ewing sarcoma can occur in almost every bone or soft tissue. Here we report a primary expansile bony lesion in the left femur mimicking Ewing’s sarcoma which on extensive workup was diagnosed as metastasis of adrenal neuroblastoma. This is the first report of this kind in the literature.
Case Report: A 3-year-old female child presented with complaints of swelling in the left thigh for 2 months. She did not have any abdominal complaints. Her initial X-ray of the thigh revealed increased density and thickness of soft tissue in the mid and distal thigh and laminated periosteal reaction with focal areas of sunburst appearance in the left femoral diaphysis region. Ultrasound (USG) of the thigh confirmed the findings of the X-ray and a provisional diagnosis of Ewing’s sarcoma was considered. During the routine workup, an USG abdomen was done which revealed an infiltrating retroperitoneal mass crossing the midline, encasing the aorta and its branches. Contrast-enhanced computed tomography of the abdomen confirmed the sonographic findings that a heterogeneously enhancing mass encasing and displacing the abdominal aorta and the left kidney was noted. The left adrenal gland was not separately visualized. Based on these findings, the primary diagnosis of metastatic neuroblastoma bony metastasis was made. Histopathology and immuno-histochemistry of the left suprarenal mass confirmed the diagnosis of neuroblastoma.
Conclusion: Neuroblastoma can primarily present with musculoskeletal symptoms such as expansile metastatic bony lesions or pathological fractures and should be considered in the differential diagnosis.
Keywords: Neuroblastoma, neuroblastoma bony metastasis, Ewing sarcoma.
Soft-tissue thigh swelling is not an uncommon presentation in children. Associated clinical features of pain, fever, and joint involvement must be evaluated in these cases. Neuroblastoma is an embryonic tumor of the peripheral sympathetic nervous system. It is the most common extracranial solid tumor of childhood and accounts for up to 15% of all pediatric cancer fatalities. The manifestation of neuroblastoma is variable depending on the location of the tumor and the presence or absence of paraneoplastic syndromes. Neuroblastoma has a great propensity to metastasize to multiple organs. The most common site of metastasis is the bone and bone marrow [1]. Ewing sarcoma is a rare small round blue cell tumor originating from neuroectoderm. It is the most common primary bone tumor of adolescents. The most common anatomical sites of involvement include the pelvis, axial skeleton, and femur; however, Ewing sarcoma can occur in almost every bone or soft tissue. Ewing sarcoma is the second most common primary malignant bone tumor in children and adolescents, representing about 3% of all pediatric cancers. The most affected sites are the femur, ilium, tibia, humerus, fibula, ribs, and sacrum. Clinical presentation typically involves pain, mass, or swelling, with symptoms persisting for over 6 months before diagnosis [2]. Neuroblastoma is a complex heterogenous embryonal tumor of primary neural crest cell origin. It is the 3rd most common pediatric tumor and the most common extracranial solid tumor. Neuroblastoma commonly originates in the adrenal gland (40%) and affects the paraspinal ganglia (25%), mediastinum (15%), neck (5%), and pelvis (3%). The majority of cases (60–70%) are metastatic upon diagnosis. The median age at diagnosis is 22 months, with approximately 81.5% of cases diagnosed by the age of 4, and an additional 15% diagnosed by the age of 9. Presentations range from palpable abdominal lump to hypertension, symptoms of nerve compression, bone metastasis, and raccoon eyes (due to orbital ecchymoses causing darkening of periorbital tissues). Less than 2% of the patients present with paraneoplastic syndrome such as profuse diarrhea (due to secretion of vasoactive intestinal peptide) or opsoclonus-myoclonus-ataxia [3]. To prevent further misdiagnosis and given the difficulties we faced in terms of radiological findings and clinical presentation, we present a child with a left thigh mass mimicking Ewing’s sarcoma on radiography; however, on further workup found out as metastatic suprarenal neuroblastoma with bony metastasis.
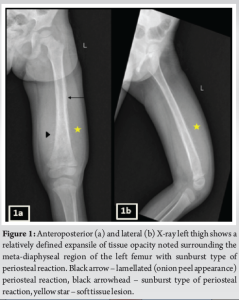
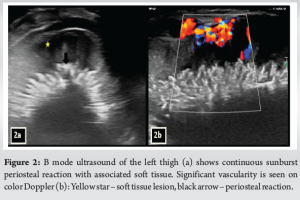
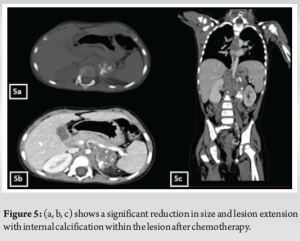
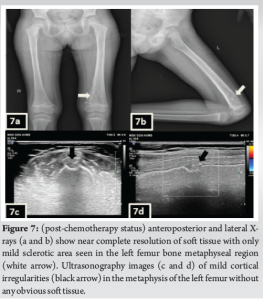
A 3-year-old female was brought to the pediatric orthopedic department with complaints of swelling over her left leg for 2 months. The swelling was insidious in onset, progressive in nature, associated with pain which was aggravated by movement, more at night, and associated with fever and local rise of temperature. According to the mother, weight loss and loss of appetite were noted. No history of abdominal pain. An X-ray of the left thigh was requested. It revealed an ill-defined subtle expansile lytic lesion in the diaphysis and distal metaphysis region of the left femur with the suggestion of lamellated (onion peel) and sunburst (in a few areas) types of periosteal reaction (Fig. 1). No evidence of fracture was noted. Laboratory results were as follows; white blood cell – 7220 cells/μL, hemoglobin – 6.8 g/dL (decreased), hematocrit – 24%, platelets – 190,000/μL, C-reactive protein (CRP) – 81.5 mg/L (raised) and lactate dehydrogenase (LDH) – 467.99 U/L (raised). The differential diagnosis of primary bone tumor (Ewing’s sarcoma > osteosarcoma) and osteomyelitis was considered initially. (High Resolution Sonography)left thigh was done, which revealed soft tissue showing significant vascularity on color Doppler with associated spiculated sunburst type of periosteal reaction (Fig. 2). A provisional diagnosis of primary bone tumor was made and as a part of metastatic screening, ultrasonography of the abdomen was advised. It revealed a large ill-defined heterogeneous retroperitoneal mass crossing the midline, encasing the aorta and its branches. The mass was seen insinuating beneath the aorta, lifting it off the vertebral column. Multiple enlarged pre/para-aortic lymph nodes and bilateral common, external, and internal iliac vessels were noted (Fig. 3). To further characterize the abdominal mass, the contrast-enhanced computed tomography (CECT) chest with the abdomen covering the thigh was done. It revealed a large heterogeneous solid mass lesion epicenter in the left suprarenal region measuring 10 × 6.6 × 11.6 cm in maximum orthogonal dimensions and crossing the midline. It shows multiple amorphous and chunky calcifications within. The mass is heterogeneously enhanced with areas of necrosis and is encasing and displacing the abdominal aorta. The left adrenal gland was not seen separately and mass is seen displacing the left kidney inferiorly and laterally. Multiple enlarged heterogeneously enhancing retroperitoneal lymph nodes are seen along with bony metastatic lesions (Fig. 4). Based on these findings, the primary diagnosis of left suprarenal neuroblastoma with multiple lymph-nodal and bony metastasis was made. Ultrasound (USG)-guided tru-cut biopsy of the left suprarenal mass was performed, which revealed small round blue cell tumor cells arranged in sheets and nests. Bone marrow trephine biopsy taken from bilateral posterior superior iliac spine revealed metastatic deposits of small round blue cell tumor. These cells were positive for synaptophysin, chromogranin, and cluster of differentiation-99 and negative for leukocyte common antigen confirming the diagnosis of neuroblastoma (Fig. 6). Eight cycles of neo-adjuvant chemotherapy were given to the patient. A metaiodobenzylguanidine scan post-chemotherapy revealed no residual bony metastasis. Positron emission tomography-computed tomography (PET-CT) was also done which revealed a non-fludeoxyglucose tissue mass noted in the left suprarenal region with an unremarkable musculoskeletal system. Follow-up CECT abdomen with the chest was done to assess chemotherapy response and for pre-operative planning which revealed a significant reduction in size and extent of the lesion with a significant reduction in number and size of retroperitoneal lymph nodes (Fig.5,7). Surgery was done and the residual lesion was removed. The patient is now on follow-up.



Thigh swelling is a common complaint in children. The onset and progression of swelling, history of fever, and pain must be assessed. Differentials to be considered include infective/inflammatory causes such as osteomyelitis, metastatic neuroblastoma, primary bone tumors such as Ewing’s sarcoma, osteosarcoma, and soft-tissue tumors such as angiosarcoma, synovial sarcoma, malignant melanoma, melanotic neuroectodermal tumors of infancy, granulocytic sarcoma, and malignant lymphoma. High CRP and LDH levels and moderate anemia suggest malignancy more than infection. In children <5 years old, osteosarcoma and Ewing’s sarcoma are primarily considered. Other rare tumors such as neuroblastoma and Wilms tumor need to be considered in under 5 children [3]. A suggested imaging protocol for the child presenting with a soft-tissue mass following clinical assessment is a plain radiograph and USG examination with color Doppler to assess a lesion’s vascularity. In many instances, a firm or provisional diagnosis is possible with these investigations. If the diagnosis is unclear from these investigations, then magnetic resonance imaging (MRI) or computed tomography (CT) are indicated. The full extent of any lesion may require an MRI or CT to demonstrate the anatomical relationships for surgical planning. An expansile lytic lesion with a periosteal reaction involving diaphysis of long bones is commonly thought of in terms of Ewing’s sarcoma and osteomyelitis [4]. Even metastasis from common pediatric tumors such as neuroblastoma should also be considered in such cases. The presence of soft-tissue mass and interrupted type of periosteal reaction favors neoplastic etiology more than osteomyelitis. Soft-tissue components are better interpreted on high-resolution USG. If the USG examination is not conclusive or cannot completely assess the lesion, MRI is often used as a second-line imaging modality [5]. Ewing sarcoma is the second most common primary malignant bone tumor in children and adolescents, representing about 3% of all pediatric cancers. It predominantly affects individuals between the ages of 4 and 25, with a peak prevalence between 10 and 15 years. There is a slight male predilection, and it is more prevalent in white patients [6]. Clinical presentation typically involves pain, mass, or swelling, with symptoms persisting for over 6 months before diagnosis. Fever and increased erythrocyte sedimentation rate are also observed in a significant number of patients. The most commonly affected sites are the femur, ilium, tibia, humerus, fibula, ribs, and sacrum. Radiographs frequently reveal a diaphyseal lesion in the diaphyseal region with onion skin periosteal reaction [7]. Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma are a spectrum of tumors arising from neural crest cells or primitive sympathetic ganglion cells. Overall, approximately 46% of them arise from adrenals and 18% from extraadrenal abdominal locations 14% from the posterior mediastinum, and the rest from neck/pelvis/other locations. It accounts for 15% of the fatalities due to its aggressive nature and metastatic disease at presentation. Approximately 41% of neuroblastoma cases are detected within the first 3 months of life, and the median age at diagnosis is 19 months. The prognosis of neuroblastoma is influenced by age, with children under 1 year old having a significantly higher 5-year survival rate compared to those diagnosed later. There are racial and ethnic disparities in both the risk and survival rates of children with neuroblastoma [8]. Neuroblastoma is commonly intra-abdominal location with lung invasion through lymphatic circulation and bone marrow, bone, skin, and liver via hematogenous circulation. Diffuse bone marrow and bone involvement can cause claudication, pain, discomfort, pathological fracture, and cytopenia. Imaging modalities ranging from radiographs to PET and bone scans are used for complete evaluation, staging, and management accordingly. The prognosis depends on the age of the patient at diagnosis. Primary involvement of the musculoskeletal system occurs only in 15% of neuroblastoma cases [9]. Small round blue cell tumors reported in histopathology reports are seen in neuroblastoma, Ewing’s sarcoma, rhabdomyosarcoma, non-Hodgkin lymphoma, and primitive neuro-ectodermal tumors [10]. In our case, the primary presentation was a mass involving the left femur which mimicked Ewing’s sarcoma on X-ray; however, on abdominal USG, and CECT abdomen, an imaging diagnosis of neuroblastoma with bony metastasis was made, which was confirmed on histopathology.
This case demonstrates the challenges in the diagnosis of metastatic neuroblastoma with initial misdiagnosis, and prolonged work-up. Radiographic appearance may be misleading and can contribute to delayed diagnosis. Bony metastasis of neuroblastoma should be considered in the differential diagnosis of any child presenting with expansile bony lesions or pathological fractures even without known primary.
Metastatic neuroblastoma as a swelling in the extremities can masquerade as a primary bone tumor such as Ewing’s sarcoma in radiography. Hence, before taking a radical surgical option, a detailed clinical examination and clinical work-up should be done to exclude any primary cause.
References
- 1.Kembhavi SA, Shah S, Rangarajan V, Qureshi S, Popat P, Kurkure P. Imaging in neuroblastoma: An update. Indian J Radiol Imaging 2015;25:129-36. [Google Scholar | PubMed]
- 2.Murphey MD, Senchak LT, Mambalam PK, Logie CI, Klassen-Fischer MK, Kransdorf MJ. From the radiologic pathology archives: Ewing sarcoma family of tumors: radiologic-pathologic correlation. Radiographics 2013;33:803-31. [Google Scholar | PubMed]
- 3.Claps G, Faouzi S, Quidville V, Chehade F, Shen S, Vagner S, et al. The multiple roles of LDH in cancer. Nat Rev Clin Oncol 2022;19:749-62. [Google Scholar | PubMed]
- 4.Henninger B, Glodny B, Rudisch A, Trieb T, Loizides A, Putzer D, et al. Ewing sarcoma versus osteomyelitis: Differential diagnosis with magnetic resonance imaging. Skeletal Radiol 2013;42:1097-4. [Google Scholar | PubMed]
- 5.Pope T. Musculoskeletal Imaging. 2nd ed. Oxford: OUP Oxford; n.d.2003. [Google Scholar | PubMed]
- 6.Muralidhar D, Vasugi GA, Sundaram S, Vasugi A. Incidence and demographic profile of Ewings sarcoma: Experience from a tertiary care Hospital. Cureus 2021;13:e18339. [Google Scholar | PubMed]
- 7.Zöllner SK, Amatruda JF, Bauer S, Collaud S, De Álava E, DuBois SG, et al. Ewing sarcoma-diagnosis, treatment, clinical challenges and future perspectives. J Clin Med 2021;10:1685. [Google Scholar | PubMed]
- 8.Choi JH, Ro JY. Mediastinal neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: Pathology review and diagnostic approach. Semin Diagn Pathol 2022;39:120-30. [Google Scholar | PubMed]
- 9.Liu S, Yin W, Lin Y, Huang S, Xue S, Sun G, et al. Metastasis pattern and prognosis in children with neuroblastoma. World J Surg Oncol 2023;21:130. [Google Scholar | PubMed]
- 10.Gregorio A, Corrias MV, Castriconi R, Dondero A, Mosconi M, Gambini C, et al. Small round blue cell tumours: Diagnostic and prognostic usefulness of the expression of B7‐H3 surface molecule. Histopathology 2008;53:73-80. [Google Scholar | PubMed]






Related Articles in Journal of Orthopaedic Case Reports
 April 1, 2025 Unveiling the Uncommon: Ewing Sarcoma Cranium – A Rare Clinical Vignette
April 1, 2025 Unveiling the Uncommon: Ewing Sarcoma Cranium – A Rare Clinical Vignette July 1, 2025 Acute Onset Common Peroneal Nerve Palsy Secondary to Fibular Head Osteochondroma in an Adolescent: A Case Report
July 1, 2025 Acute Onset Common Peroneal Nerve Palsy Secondary to Fibular Head Osteochondroma in an Adolescent: A Case Report September 1, 2024 Bone Preservation in Femoral Intercondylar Box Cut – A Comparative Study between Older and Newer Generation Implants
September 1, 2024 Bone Preservation in Femoral Intercondylar Box Cut – A Comparative Study between Older and Newer Generation Implants April 10, 2021 Arthroscopic Removal of Tenosynovial Giant-Cell Tumors of the Cruciate Ligaments. Presentation of Two Cases
April 10, 2021 Arthroscopic Removal of Tenosynovial Giant-Cell Tumors of the Cruciate Ligaments. Presentation of Two Cases