[box type=”bio”] What to Learn from this Article?[/box]
RCDP can present in paediatric age group. Management is symptomatic and supportive. Physiotherapists and occupational therapists can help relieve the symptoms of a child’s unusual skeletal development and cataract can be removed surgically.
Case Report | Volume 5 | Issue 3 | JOCR July-Sep 2015 | Page 38-40 | Yashwant Mahale, Vikram V. Kadu, Amit Chaudhari . DOI: 10.13107/jocr.2250-0685.303.
Authors: Yashwant Mahale[1], Vikram V Kadu[1], Amit Chaudhari[1]
[1] Department of Orthopaedics, ACPM Medical College, Dhule, Maharashtra, India.
Address of Correspondence
Dr. Vikram Vilasrao Kadu
C/O Vilas Shamrao Kadu, Plot no. 20, Kadu House, Barde layout, Friends colony, Katol Road, Nagpur – 440013. Maharashtra. India.
E mail – vikram1065@gmail.com
Abstract
Introduction: Introduction: Rhizomelic chondrodysplasia punctata (RCDP) is a very rare disease. It impairs the normal development of many parts of the body. The features of this disorder include bony abnormalities, severe mental retardation, joint contractures, cataract and recurrent respiratory infections and breathing problems. Seizures and Distinctive facial features including prominent forehead, depressed nasal bridge and small nose is also associated with this pathology. Being rare, this is very difficult to diagnose when presented at OPD. Proper history and meticulous examination is extremely necessary. Our aim is to discuss current knowledge on etiopathogenesis as well as radiological and clinical symptoms of diseases associated with RCDP.
Case Report: 5 yrs old male child presented with chest infection and periarticular swelling of all the small and large joints. The patient was walking with limp. History elicited that the child was born of a consanguineous marriage. The child was delivered at home. Birth weight was 2.4 kgs. He repeatedly had upper respiratory tract infections and was taking treatment for the same. He was further investigated in the form of clinical, biochemical and radiological assessment which stated that the patient was suffering from RCDP.
Conclusion: This is a rare presentation. Though this is not curable, management of RCDP is symptomatic and supportive and may include physiotherapy and orthopedic procedures (in later stages) to improve function. The child may also undergo cataract surgery to improve vision.
Keywords: RCDP, Paediatric, PEX7.
Introduction
Rhizomelic chondrodysplasia punctata type 1 (RCDP1) is an inherited disease characterized by skeletal abnormalities, growth retardation, severe mental retardation, cataracts and has decreased life expectancy [1]. It impairs the normal development of many parts of the body. These affected individuals have a specific bone abnormality called chondrodysplasia punctata, which shows scattered calcifications at the end of the long bones thus affecting their growth. People with rhizomelic chondrodysplasia punctata often develop joint deformities (contractures) that makes the joints stiff and painful. This condition is considered as a developmental brain disorder characterized by systemic shortening of the proximal bones (i.e. rhizomelia), seizures, recurrent respiratory tract infections, and congenital cataracts. 3 genetic subtypes has been reported of which RCDP type 1, caused by mutations in the Peroxisomal biogenesis factor 7 gene, is the most common type. RCDP type 2 and 3 has single enzyme deficiencies in the plasmalogen biosynthesis pathway.
Case report
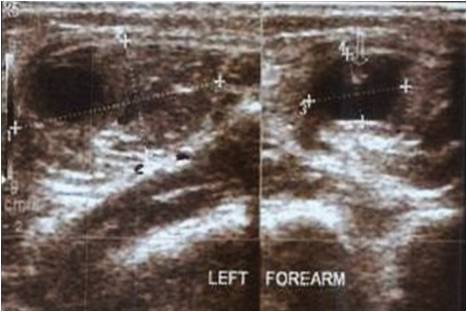
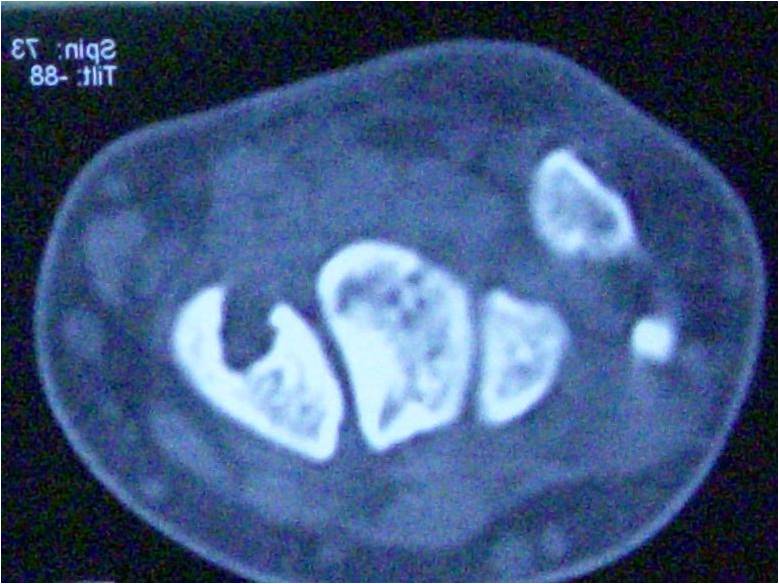
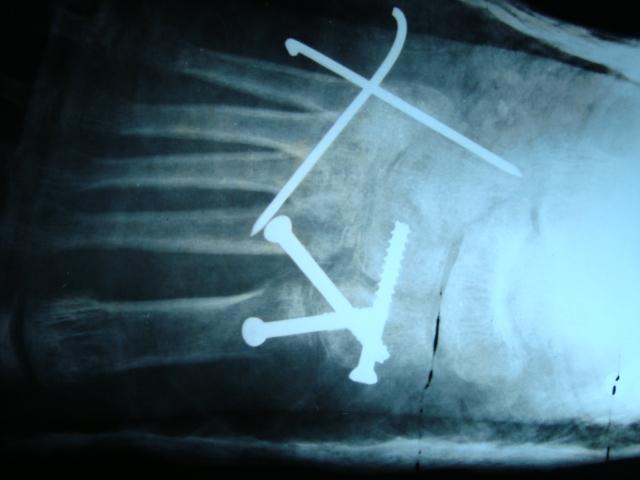
A five years male child presented to the paediatric OPD with complaints of recurrent upper respiratory tract infection. On examination he had swelling of the joints for which the child was referred to our OPD. Examination revealed that the swelling was non-tender, bony hard and did not show any effusion. Thorough examination with proper history taking was done which revealed that the child was born of a 3rd degree consanguineous marriage at home after 38 weeks of gestation from the third pregnancy of a healthy 26-year-old mother and 30 yrs old father. The mother was under routine prenatal follow-up during pregnancy. There was no history of intake of toxic drug or any chronic medications. The mother had two deliveries, the first two children being normal. Birth Weight of this child was 2.4 kgs. Currently at 5 yrs of age, weight is 10.2 kgs, Length of Upper extremities (41cms) and that of lower extremities (36.5 cms), height is 81 cms, head circumference is 47 cms, chest circumference is 49 cms. The patient had history of upper respiratory tract infection frequently (once a month or once in 2 months). Clinically, the child had congenital cataract in left eye and early cataract changes in Right eye (Fig 1). Facial features showed prominent forehead, widely set eyes and a sunken face (Fig 2). The patient can speak sentence, can run, can dress and undress himself. At 1.5 yrs of age he started sitting without support and speaking bisyllables. The child was walking with limp. All Milestones were achieved till date. Weight by age and height by age was less than 3rd percentile (Fig 4). Normal weight should have been around 18 kgs (weight is 10.2 kgs) and normal height around 115 cms (height is 81cms). The radiological findings of the patient were compatible with CDP with punctate calcifications in the epiphyses and metaphyseal dysplasia (Fig 5,6,7). Blood investigations revealed Alkaline phosphatase – 160, serum Ca+ – 9.2, Sr. phosphorous – 5, ESR – 130. RCDP was diagnosed based on clinical and radiological criteria.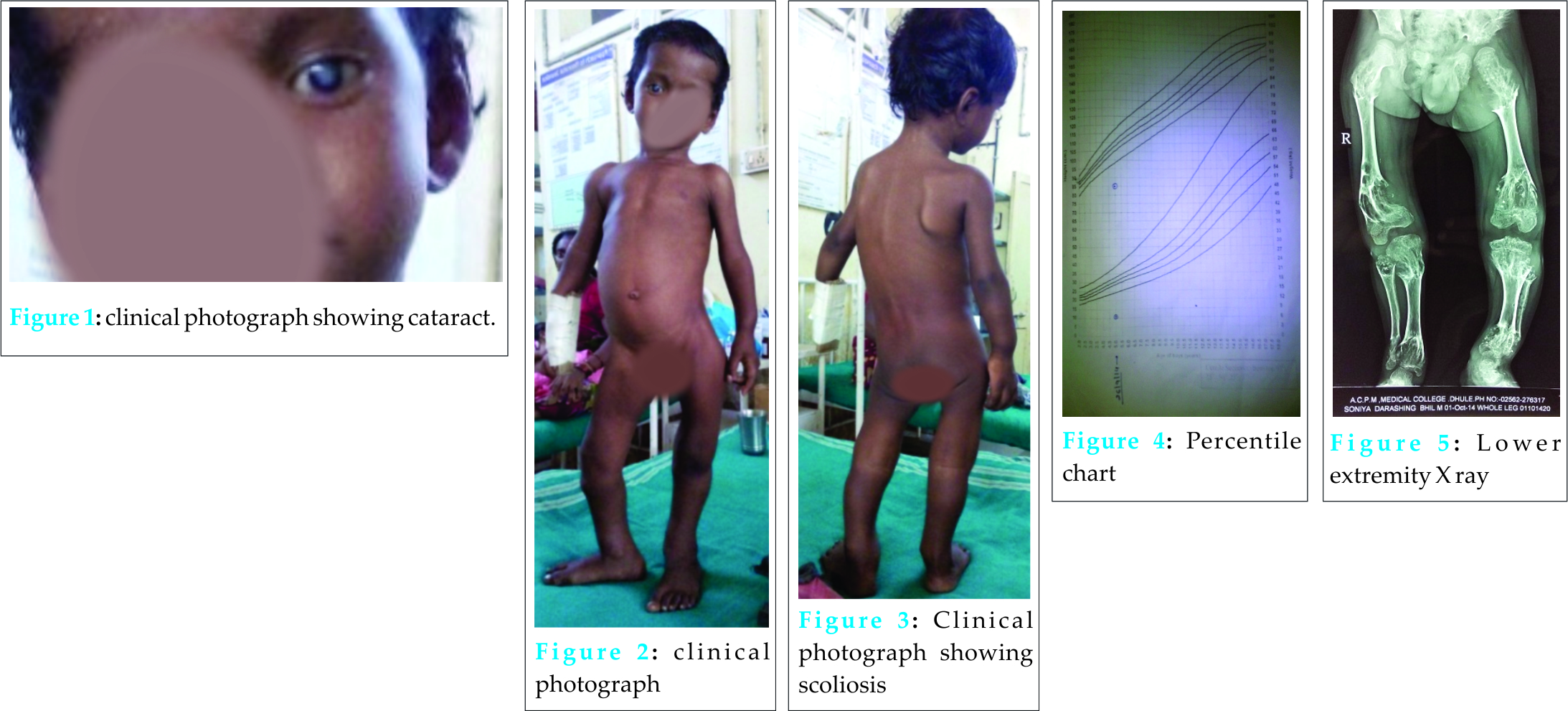
Discussion
Rhizomelic chondrodysplasia punctata type 1 is an inherited disease with extremely rare presentation. It affects 1 in 100,000 individuals [2]. Rhizomelic chondrodysplasia punctata is associated with significantly delayed development and severe mental retardation. Most children with this condition do not achieve developmental milestones and has growth retardation. These children may also have seizure episodes. It is generally associated with recurrent respiratory infections and breathing problems which may be life threatening. Because of their severe health problems, most children with this do not survive long. It is rare for affected child to live past age 10. The milder form of this disease is characterized milder degrees of rhizomelia along with growth and mental retardation. Distinctive facial features such as prominent forehead, widely set eyes, sunken appearance of face, small nose. Additionally, almost all affected individuals present with cataract. The cataracts are apparent at birth (congenital) or develop in early infancy.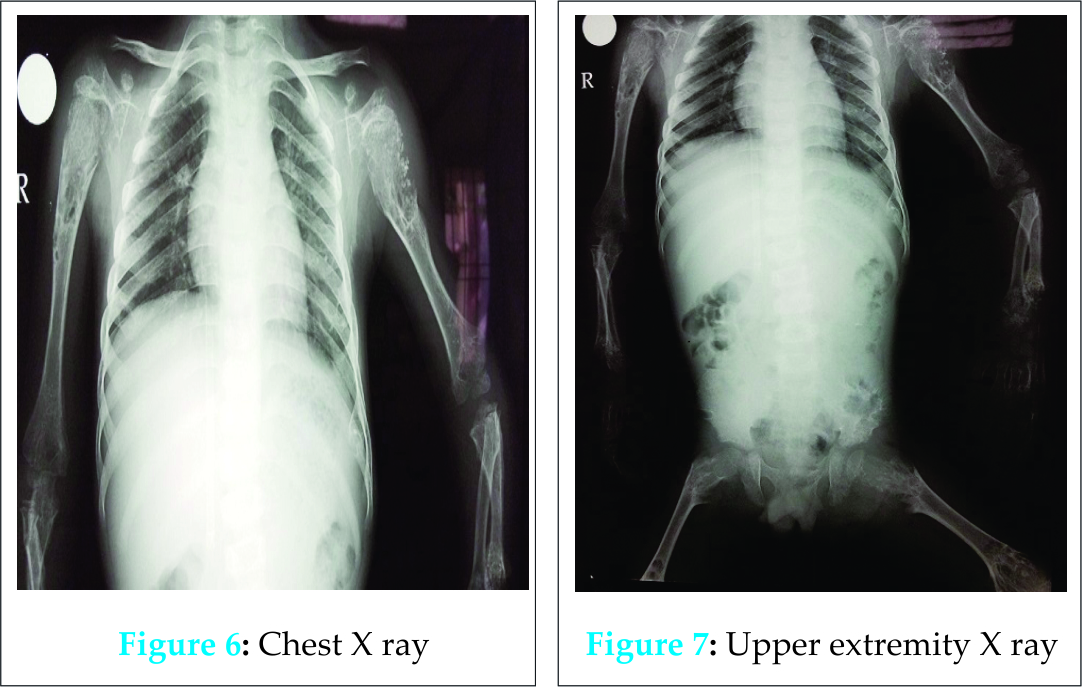
RCDP is an autosomal-recessive disease caused by mutations in the PEX7 gene. An individual who inherits one copy of a PEX7 gene mutation is a “carrier” and do not have related health problems. An individual who inherits mutations from each parent, is expected to be affected with RCDP. Thus it shows that RCDP is an inherited disease but results only when the genes are inherited from both the parents.
If mother and father both are carriers, the chances of getting affected for a child is 25% in each pregnancy; therefore, it is of utmost importance that the reproductive partner of a carrier be offered testing. A negative does not eliminate the possibility of inheritance of the gene mutation by the child.
Conclusion
RCDP is a rare presentation in paediatric age group. There is no cure for RCDP. Unfortunately, most children do not survive after age of 10. Diagnosis was done mainly on the basis of clinical examination and investigations such as X-rays. We concluded that, Management of RCDP is symptomatic and supportive and may include physiotherapy and orthopedic procedures (in later stages) to improve function. The child may also undergo cataract surgery to improve vision.
Clinical Message
Though very rare, RCDP can present in paediatric age group. Although there is no cure for RCDP, management is symptomatic and supportive. Physiotherapists and occupational therapists can help relieve the symptoms of a child’s unusual skeletal development and cataract can be removed surgically.
Reference
1. Braverman NE, et al. Rhizomelic chondrodysplasia punctata type 1. Gene Reviews NCBI Bookshelf National Library of Medicine, NIH Available at: http://www.ncbi.nlm.nih.gov/books/NBK1270. Accessed February 22, 2012.
2. Genetics Home Reference. Rhizomelic chondrodysplasia punctata. Available at: http://ghr.nlm.nih.gov/condition/rhizomelic-chondrodysplasia-punctata. Accessed March 1, 2012.
3. Weller, S.; Gould, S. J.; Valle, D. (2003). “Peroxisome Biogenesis Disorders”. Annual Review of Genomics and Human Genetics 4: 165–211.doi:10.1146/annurev.genom.4.070802.110424. PMID 14527301
4. A. M. Bams-Mengerink, J. H. Koelman, H. Waterham, P. G. Barth, and B. T. Poll-The, “The neurology of rhizomelic chondrodysplasia punctata,” Orphanet Journal of Rare Diseases, vol. 8, no. 1, article 174, 2013. View at Publisher · View at Google Scholar
5. M. D. Irving, L. S. Chitty, S. Mansour, and C. M. Hall, “Chondrodysplasia punctata: a clinical diagnostic and radiological review,” Clinical Dysmorphology, vol. 17, no. 4, pp. 229–241, 2008. View at Publisher· View at Google Scholar · View at Scopus
6. C. T. Yalin, I. K. Bayrak, M. Danaci, and L. Incesu, “Case report: rhizomelic chondrodysplasia punctata and foramen magnum stenosis in a newborn,” Turkish Journal of Diagnostic and Interventional Radiology, vol. 9, no. 1, pp. 100–103, 2003. View at Scopus
7. S. C. Morrison, “Punctate epiphyses associated with Turner syndrome,” Pediatric Radiology, vol. 29, no. 6, pp. 478–480, 1999. View at Publisher · View at Google Scholar · View at Scopus
8. A. Leicher-Duber, R. Schumacher, and J. Spranger, “Stippled epiphyses in fetal alcohol syndrome,”Pediatric Radiology, vol. 20, no. 5, pp. 369–370, 1990. View at Scopus
9. J.-L. D. Alessandri, D. Ramful, and F. Cuillier, “Binder phenotype and brachytelephalangic chondrodysplasia punctata secondary to maternal vitamin K deficiency,” Clinical Dysmorphology, vol. 19, no. 2, pp. 85–87, 2010. View at Publisher · View at Google Scholar · View at Scopus
10. A. K. Poznanski, “Punctate epiphyses: a radiological sign not a disease,” Pediatric Radiology, vol. 24, no. 6, pp. 418–424, 1994. View at Publisher · View at Google Scholar · View at Scopus
11. A. L. White, P. Modaff, F. Holland-Morris, and R. M. Pauli, “Natural history of rhizomelic chondrodysplasia punctata,” American Journal of Medical Genetics, vol. 118, no. 4, pp. 332–342, 2003.View at Scopus
12. A. L. Shanske, L. Bernstein, and R. Herzog, “Chondrodysplasia punctata and maternal autoimmune disease: a new case and review of the literature,” Pediatrics, vol. 120, no. 2, pp. e436–e441, 2007. View at Publisher · View at Google Scholar · View at Scopus
13. I. Singh, G. H. Johnson, and F. R. Brown III, “Peroxisomal disorders. Biochemical and clinical diagnostic considerations,” American Journal of Diseases of Children, vol. 142, no. 12, pp. 1297–1301, 1988.View at Scopus
14. P. Violas, B. Fraisse, M. Chapuis, and H. Bracq, “Cervical spine stenosis in chondrodysplasia punctata,”Journal of Pediatric Orthopaedics B, vol. 16, no. 6, pp. 443–445, 2007. View at Publisher · View at Google Scholar · View at Scopus
15. T. E. Herman, B. C. P. Lee, and W. H. McAlister, “Brachytelephalangic chondrodysplasia punctata with marked cervical stenosis and cord compression: report of two cases,” Pediatric Radiology, vol. 32, no. 6, pp. 452–456, 2002. View at Publisher · View at Google Scholar · View at Scopus
16. E. Jurkiewicz, B. Marcinska, J. Bothur-Nowacka, and A. Dobrzanska, “Clinical and punctate and review of available literature,” Polish Journal of Radiology, vol. 78, no. 2, pp. 57–64, 2013. View at Publisher ·View at Google Scholar
17. J. Khanna, N. E. Braverman, D. Valle, and P. D. Sponseller, “Cervical stenosis secondary to rhizomelic chondrodysplasia punctata: brief clinical report,” American Journal of Medical Genetics, vol. 99, no. 1, pp. 63–66, 2001.
18. A. M. Bams-Mengerink, C. B. L. M. Majoie, M. Duran et al., “MRI of the brain and cervical spinal cord in rhizomelic chondrodysplasia punctata,” Neurology, vol. 66, no. 6, pp. 798–803, 2006. View at Publisher · View at Google Scholar · View at Scopus.
| How to Cite This Article: Mahale Y, Kadu VV, Chaudhari A. Rare Case of Rhizomelic Chondrodysplasia Punctata. Journal of Orthopaedic Case Reports 2015 July – Sep;5(3): 38-40. Available from: https://www.jocr.co.in/wp/2015/07/10/2250-0685-303-fulltext/ |

[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com