[box type=”bio”] What to Learn from this Article?[/box]
Use of Technology in form of 3-D printing to plan and execute complex bony surgeries.
Case Report | Volume 5 | Issue 1 | JOCR Jan-March 2015 | Page 26-30 | Achmad Fauzi Kamal, Robin Novriansyah, Rahyussalim, Yogi Prabowo, Nurjati Chairani Siregar. DOI: 10.13107/jocr.2250-0685.248
Authors: Achmad Fauzi Kamal, Robin Novriansyah, Rahyussalim, Yogi Prabowo, Nurjati Chairani Siregar
[1] Department of Orthopaedic and Traumatology RSCM, Jl. Diponegoro 71, Jakarta Pusat, Jakarta, Indonesia
[2] Department of Pathology Anatomy RSCM, Jl. Diponegoro 71, Jakarta Pusat, Jakarta, Indonesia.
Address of Correspondence:
Dr. Achmad Fauzi Kamal,
Department Orthopaedic and Traumatology, RSCM, Jl. Diponegoro 71, Jakarta Pusat, Jakarta, Indonesia, 10000. Email: fauzikamal@yahoo.com
Abstract
Introduction: Fibrodysplasia ossificans progressiva (FOP) is a rare autosomal dominant genetic disorder and characterized by postnatal progressive heterotopic ossification of the connective tissue. There are difficulties in diagnosing FOP, thus delayed or misdiagnosis and mismanagement is common.
3D printers have now become widely available and inexpensive, and can be used to rapidly produce life-size models based on CT scans of an individual patient. The availability of patient specific, accurate and detailed models of complex acetabular fractures can aid planning of surgical management on a patient specific basis.
Case Report: We present the diagnosis and surgical management of a 9-year old Indonesian girl with FOP. She presented with extensive involvement of cervical spine and both shoulders. Total excision of occipito-cervico-lumbar and paravertebral ossification and also exostoses at bilateral shoulder was done. At three years follow up, she had local recurrence with similar range of movement of the shoulder and cervical spine.
Conclusion: FOP is an extremely rare case. It is difficult to diagnose and manage FOP, therefore delayed or misdiagnosis and also inappropriate management is common.
Keywords: fibrodysplasia ossificans progressiva, heterotopic ossification.
Introduction
Fibrodysplasia ossificans progressiva (FOP), once called myositis ossificans progressiva, is a rare autosomal disorder that involves connective tissue. FOP is hall-marked by progressive heterotopic ossification and distinctive skeletal malformation. It was first reported by Patin (1692) and Freke (1739). One of the famous case of FOP is Harry Eastlack (1933 – 1973), who died because of pneumonia and fully ossified body, his lips were the only body part that could be used properly [1,2].
FOP is sporadic and not influenced by race, gender, or geographic distribution. Its incidence is 1 per 2 million people. Most patients did not have a long survival. However, some reported case could reach the old age. The average age of patients is 28.7 year old [3,4].
Signs of ossification start to appear in childhood. Abnormality of great toe is the most common finding and often noticeable at birth[5]. Other signs such as vertebral abnormality are also quite common. Ectopic ossification is preceded by a soft tissue enlargement that may spontaneously resolve or gradually form an ectopic bone. Delayed or missed diagnosis is common, which leads to inappropriate management [1,3].
We report a rare case of FOP arising at occipito-cervical spine, paravertebrae, and bilateral shoulders focusing on diagnostic procedure and surgical management. Our aim is to get a clearer view on this FOP case.
Case report
A 9-year-old girl presented to our orthopaedic clinic, with main complaint of a mass which was getting bigger on her upper back. Her parents noticed that she has already had the mass since six years. The initial mass size was around 1 cm in diameter (like a marble) located just below the tip of both scapulae. Subsequently, more masses appeared on the left lateral side of vertebrae. It was accompanied by difficulty in moving both shoulders and neck since 4 years. Because of these complains, her family tried various traditional medicines without improvement.
Six months prior to admission, she fell down on her back, and since then the masses grew progressively. She also felt pain around the masses. Meanwhile her neck and shoulder movement were becoming more limited than usual. She was brought to a district hospital in other province and then referred to our hospital to get more appropriate treatment. There was no history of similar disease in her family.
On physical examination, general condition was good and no abnormality was found in other organ. We found multiple bony prominences measuring about 2 cm in diameter at paravertebral region from cervical to lumbar and posterior thoracic wall. They made a bone bridges starting from inferior occipital bone to cervical spine and extending to paravertebral lumbar region. We also found multiple masses at both axilla. Cervical lordosis and thoracic kyphosis were diminished. All the masses were hard in consistency. There was no tenderness, and the masses were fixed to the bone.The shoulder, cervical, and thoracal spine ranges of movement were limited (Fig 1).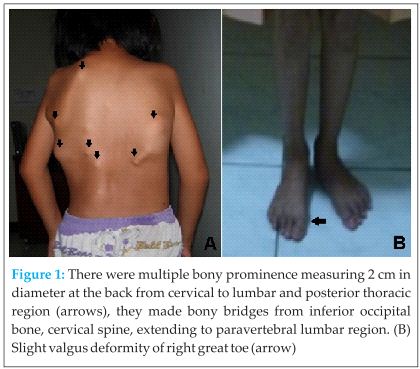
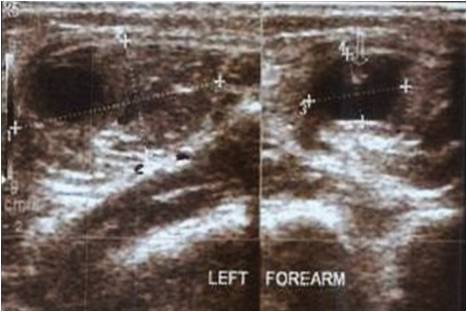
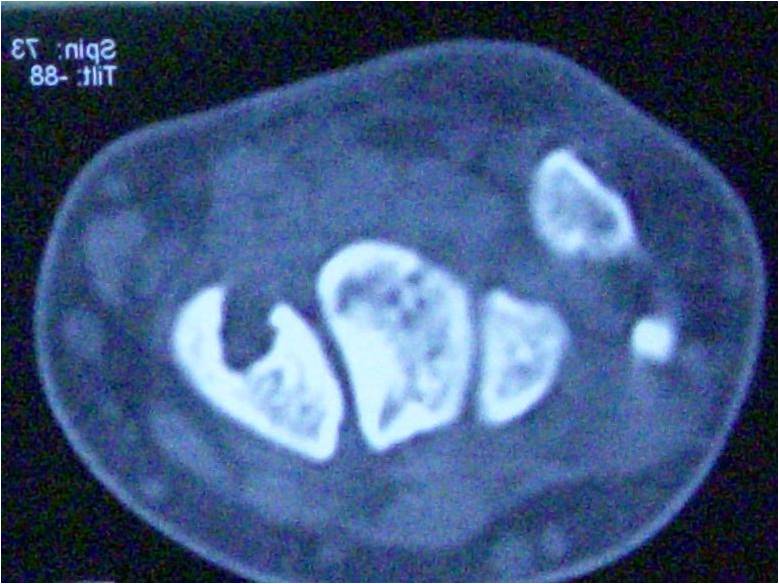
Radiograph of the cervical spine showed new bone formation starting from occipital bone, cervical, until upper thoracal spine. Chest and shoulder radiographs also showed new bone formation at both humerus bridging to the tip of scapulae, with subluxation of both humeral head superiorly. The radiologist saw it as mustiple exostoses (osteochondroma lesion) with the differential diagnosis progressive myositis ossificans. HLA-B27 was negative, it was investigated since we also thought ankylosing spondylitis as a differential diagnosis. The 3D computerized tomography (CT) reconstruction revealed multifocal ossification on soft tissue including right pectoral muscle, bilateral latissimus dorsi, and left longisimus thoracis muscle. It was in accordance with myositis ossificans progresiva (Fig 2).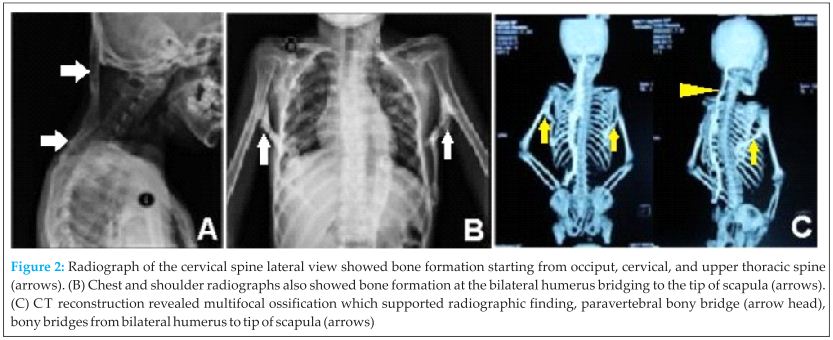 Magnetic resonance imaging (MRI) showed similar result with CT scan and no other abnormality detected at the spine.
Magnetic resonance imaging (MRI) showed similar result with CT scan and no other abnormality detected at the spine.
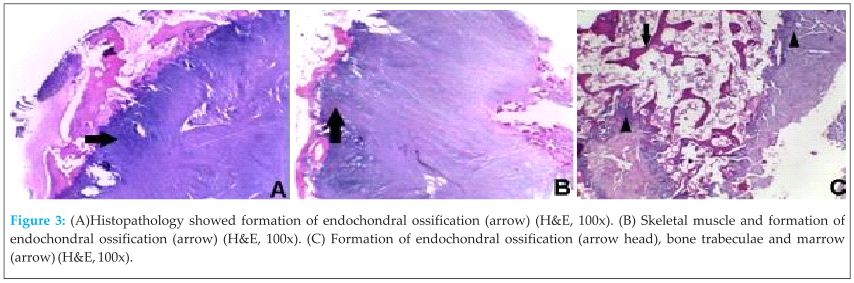
Because of the problem was multiple exostoses and stiffness, total excision of occipito-cervico-lumbar and paravertebral ossification and also exostoses at bilateral shoulder was done. Histopathological findings showed tissue consisting of cartilaginous component, irregular underlying bony trabeculae, and bone marrow between trabeculae. There was also endochondral ossification with no cellular atypia (Fig 3).
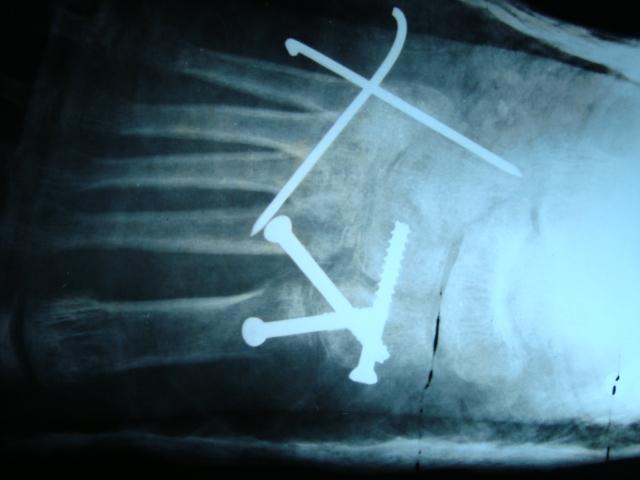
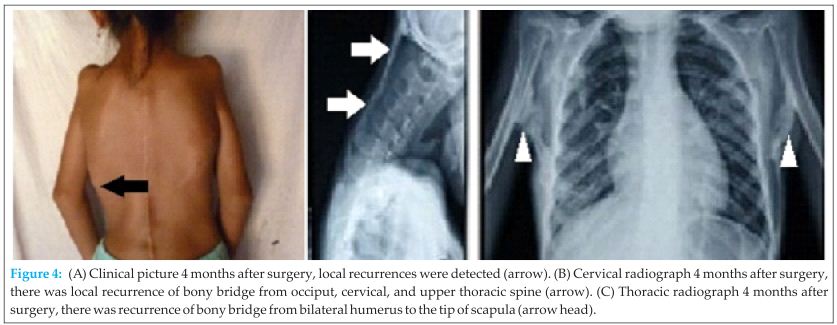
 After review of all patient’s data in one of the clinicopathological conference, we concluded that it was fibrodysplasia ossificans progressiva (FOP). Four months after surgery local recurrences were detected, which was confirmed with radiograph (Fig 4). At three years follow up, she had similar signs and symptoms as before surgery.
After review of all patient’s data in one of the clinicopathological conference, we concluded that it was fibrodysplasia ossificans progressiva (FOP). Four months after surgery local recurrences were detected, which was confirmed with radiograph (Fig 4). At three years follow up, she had similar signs and symptoms as before surgery.
Discussion
FOP is an autosomal dominant disorder and has a variable expression that is the most catastrophic disorder of heterotopic ossification in humans. Primary genetic defects in FOP are currently unknown. Events which may precipitate formation of lesion are trauma, invasive medical procedure, muscle fatigue, influenza like infection, and immunization. However, it is still unclear how clinical events which initiate the formation of lesion ultimately lead to extraskeletal ossification. A study in Japan involving 19 FOP subjects has successfully identified gene mutation that could induce ectopic bone formation in FOP [4,6].
FOP lesion is usually triggered by soft tissue injury, predominantly in muscle, causing release of inflammatory cytokines and migratory factors [7]. Potential soluble mediators include prostaglandins (PGs), Smad1 and Smad5, stromal cell derived factor 1, and BMP-4 [8]. PGE2 has an anabolic effect on bone marrow stromal cells and may enhance osteoblastic differentiation in-vitro, presumably through the recruitment of nonadherent mesenchymal progenitor cells into adherent osteoblast precursors [7]. Elevated levels of a low-molecular-weight PG-like molecule have been detected in the plasma of patients with FOP [9].
Stromal cell derived factor 1 is a mediator of lesion formation in a mouse model of pulmonary fibrosis and may signal circulating fibrocytes to homing regions that eventually become fibrotic [10]. Heterotopic bone formation may occur in pulmonary fibrosis and the circulating fibrocytes are abundant in patient with FOP with active exacerbations. Taken together, these observations suggest a plausible mechanism for extraskeletal ossification in FOP that invokes the recruitment of circulating osteogenic cells. Whereas BMP-4 may induce ectopic ossification in animal models, it is known to be dysregulated in FOP [11,12]. BMPs, Smad1, and Smad5 are signaling molecules for osteoblastic differentiation [13,14].
Children who have FOP appear normal at birth except for congenital malformations of the great toes [5]. Typically, during the first decade of life, sporadic episodes of painful soft tissue swellings (flare-ups) occur and are commonly mistaken for tumors [3]. However, in our patient, no malformations of the great toes were clearly detected except slight valgus of right great toe.
The ossification started from occipital bone and posterior paravertebral soft tissue, which caused limitation of neck movement, whereas ossification on both humerus and scapula caused limitation of the shoulder joint movement. Eventually, the ossification process forms bridges between axial bones. This clinical course was consistent with the common symptoms and signs of FOP. Most of literature stated that ectopic ossification usually started from the cervical area, then extended down paravertebrally to lower back and progressively formed “bridges” between soft tissues [4].
FOP is very rare and we had no prior experience of a similar case. Bony projections in our case were similar to multiple exostoses, whereas the location of lesions was uncommon for that kind of “tumor”. Based on our clinical diagnosis, we performed excision of “all multiple exostoses” which were located from occipital bone until lumbar spine and the bridging bones between scapula and both bilaterally.
Histopathology showed fragmented tissue consisting of cartilaginous component and irregular underlying bony trabeculae (endochondral ossification). There was also bone marrow between trabeculae, a histopathologic feature commonly found in exostosis.At discussion in our routine FOP, we re-evaluated the slides and processed the whole specimen. Microscopically the specimen also showed fibrous tissue, skeletal muscle mixed with endochondral ossification, and foci of calcification. Then, we concluded that these features were consistent with FOP. Although histologically, a very early FOP lesions look identical to the fibroproliferative lesions of aggressive juvenile fibromatosis, in aggressive juvenile fibromatosis, the lesions do not progress beyond the connective tissue growth phase, whereas in FOP, they mature through an endochondral process to form cartilage and bone [15].
There are difficulties in diagnosing FOP, thus delayed or misdiagnosis is common. One study mentioned that nearly 90% of patients with FOP worldwide are misdiagnosed and 67% undergo dangerous and unnecessary diagnostic procedures. Tumor, fibromatosis, bunion, myositis ossificans, calcified hematoma, exostoses, and also ankylosing spondylitis [3] are considered in the differential diagnosis.
Currently, there is no effective treatment for FOP. The main principle is to avoid potential condition that may trigger ectopic ossification. Surgical therapy which aims to improve range of movement in this patient has proven to trigger further ossification [3]. In this patient, local recurrences were detected at four months after surgery. At three years follow up, she presented with the same complaint and similar range of movement of shoulder and cervical spine as before surgery.
Radiation therapy may be considered after surgery to prevent recurrence, but there is no clear evidence that radiotherapy is effective in patients with FOP, except in small number of patients [16]. Medications that have theoretical application to FOP such as leukotriene inhibitor and investigational new drugs such as signal transduction inhibitor and monoclonal targeting ACVR1 may show a promising result in FOP patient. Research in FOP has reached genetic level, but not yet in molecular level that it might produce a gene therapy [17]. So, we have to make early clinical diagnosis and the mainstay of therapy is education to the patient and family to prevent further trauma.
Conclusion
In conclusion, FOP is an extremely rare case. By far, this is the first case of FOP found and reported in Indonesia. It is difficult to diagnose and manage FOP, therefore delayed or misdiagnosis and also inappropriate management is common.
Clinical Message
Although rare, we should consider FOP as a differential diagnosis in patient complaining of multiple masses with soft tissue ossification. Early clinical suspicion with prevention of trauma remains the mainstay of therapy for FOP. We should avoid dangerous and unnecessary diagnostic procedure as well as inappropriate management which may lead to futher exacerbations..
References
1. Connor JM, Evans DAP. Fibrodysplasia ossificans progressiva: the clinical features and natural history of 34 patients. J Bone Joint Surg 1982;64:76-83.
2. Hagiwara H, Aida N, Machida J, Fujita K, Okuzumi S, Nishimura G. Contrast-enhanced MRI of an early preosseous lesion of fibrodysplasi ossificans progressiva in a 21-month-old boy. Am J Roentgenol 2003;181:1145-7.
3. Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 2005;116(5):e654-61.
4. Thickman D, Bonakdar-pour A, Clancy M, Van Orden J, Steel H. Fibrodysplasia ossificans progressiva. Am J Roentgenol 1982;139:935-41.
5. Nakashima Y, Haga N, Kitoh H, Kamizono J, Tozawa K, Katagiri T, et al. Deformity of the great toe in fibrodysplasia ossificans progressiva. J OrthopSci 2010;15(6):804-9.
6. Fukuda T, Kohda M, Kanomata K, Nojim J, Nakamura A, Kamizono J, et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem 2009;284(11):7149-56.
7. Scutt A, Bertram P. Bone marrow cells are targets for the anabolic actions of prostaglandin E2 on bone: induction of a transition from nonadherent to adherent osteoblast precursors. J Bone Miner Res 1995;10(3):474–87.
8. Levitz CL, Cohen RB, Zasloff MA. The role of prostaglandins in the pathogenesis of fibrodysplasia ossificans progressiva. Calcif Tissue Int 1992;50:387.
9. Glaser DL, Economides AN, Wang L. In vivo somatic cell gene transfer of an engineered noggin mutein prevents BMP4-induced heterotopic ossification. J Bone Joint Surg Am 2003;85-A:2332–42.
10. Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age-and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Relat Res 1994;301:243-8.
11. Kaplan FS, Glaser DL, Hebela N, Shore EM. Heterotopic ossification. J Am Acad Orthop Surg 2004;12:116–25.
12. Cunningham NS, Paralkar V, Reddi AH. Osteogenin and recombinant bone morphogenetic protein 2B are chemotactic for human monocytes and stimulate transforming growth factor beta 1 mRNA expression. Proc Natl Acad Sci USA 1992;89(24):11740–4.
13. Zvaifler NJ, Marinova-Mutafchieva L, Adams G, Edwards CJ, Moss J, Burger JA, et al. Mesenchymal precursor cells in the blood of normal individuals. Arthritis Res 2000;2:477–88.
14. Kuznetsov SA, Mankani MH, Gronthos S, Satomura K, Bianco P, Robey PG. Circulating skeletal stem cells. J Cell Biol 2001;153:1133–40.
15. Kaplan FS, Tabas J, Gannon FH, Finkel G, Hahn GV, Zasloff MA. The histopathology of fibrodysplasia ossificans progressiva: an endochondral process. J Bone Joint Surg 1993;75(2):220–230.
16. Soldic Z, Murgic J, Radic J, Dabelic N, Jazvic M, Brozic JM, et al. Radiation therapy in treatment of fibrodysplasia ossificans progressiva: a case report and review of the Literature.Coll Antropol 2011;35(2):611–4.
17. Kaplan FS, Shore EM, Pignolo RJ. The medical management of fibrodysplasia ossificans progressiva: treatment considerations. Clin Proc Intl Clin Consort FOP 2011;4:1-100.
| How to Cite This Article: Kamal AF, Novriansyah R, Rahyussalim, Prabowo Y, Siregar NC. Fibrodysplasia Ossificans Progressiva: Difficulty in Diagnosis and Management A case report and literature review. Journal of Orthopaedic Case Reports 2015 Jan-March;5(1): 26-30. Available from: https://www.jocr.co.in/wp/2015/01/28/2250-0685-248-fulltext/ |

[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com