[box type=”bio”] Learning Point of the Article: [/box]
[box type=”bio”] Learning Point of the Article: [/box]
Early genetic diagnosis and orthopedic care improves the outcome of diastropic dysplasia.
Case Report | Volume 11 | Issue 2 | JOCR February 2021 | Page 81-85 | Anchal Kumar Tripathi, Sunny Choudhary, Vivek Singh, Prashant Kumar Verma. DOI: 10.13107/jocr.2021.v11.i02.2036
Authors: Anchal Kumar Tripathi[1], Sunny Choudhary[2], Vivek Singh[2], Prashant Kumar Verma[1]
[1]Department of Pediatrics, All India Institute of medical Sciences, Rishikesh, Uttarakhand, India,
[2]Department of Orthopedic Surgery, All India Institute of medical Sciences, Rishikesh, Uttarakhand, India.
Address of Correspondence:
Dr. Prashant Kumar Verma,
Department of Pediatrics, All India Institute of medical Sciences, Rishikesh – 249 203, Uttarakhand, India.
E-mail: p.a.9457@gmail.com
Abstract
Introduction: Diastrophic dysplasia (DTD) results from SCN26A2 gene mutation, with autosomal recessive inheritance and widely variable phenotype. The gene has been mapped to chromosome 5q32-q33.1.
Case Report: We present a case of a 4-year-old female with short stature, bilateral feet and knee deformity, and dysplastic facies. SCN26A2 mutations were seen in patient as well as parents. She underwent multiple orthopedic procedures involving metatarsals, gastrosoleus, and distal femur. Based on typical clinical features, DTD was suspected. Genetic studies of patient and parents provided the exact diagnosis in this case.
Conclusion: Genetic diagnosis and family counseling are important caveat of management. Key features like ear abnormalities help to suspect diagnosis which requires a high index of suspicion. Associated bony and soft-tissue abnormalities of lower limb may require surgical intervention for improvement of gait, functions, and cosmesis.
Keywords: Diastrophic dysplasia, Skeletal dysplasia, Ear abnormalities, SCN26A2, Osteotomy.
Introduction
Bones and cartilage are the predominantly affected structures in skeletal dysplasias, various other manifestations are seen as a consequence of these dysplasias [1]. Mutations in genes encoding diastrophic dysplasia (DTD) sulfate transporter (DTDST or SLC26A2) lead to the development of a group of skeletal dysplasias that comprise the most severe phenotypic disorder called achondrogenesis 1B followed by atelosteogenesis 2 and less severe phenotypic presentations in DTD and recessive multiple epiphyseal dysplasia with possible genotype and phenotype relationship. All these disorders are autosomal recessive in inheritance patterns. The gene has been mapped to chromosome 5q32-q33.1. The protein encoded by this SLC6A2 gene acts as sulfate/chloride antiporter of the cartilage cell membrane. In turn, cartilage cells incorporate sulfate ions to manufacture proteoglycans [2].
Case Report
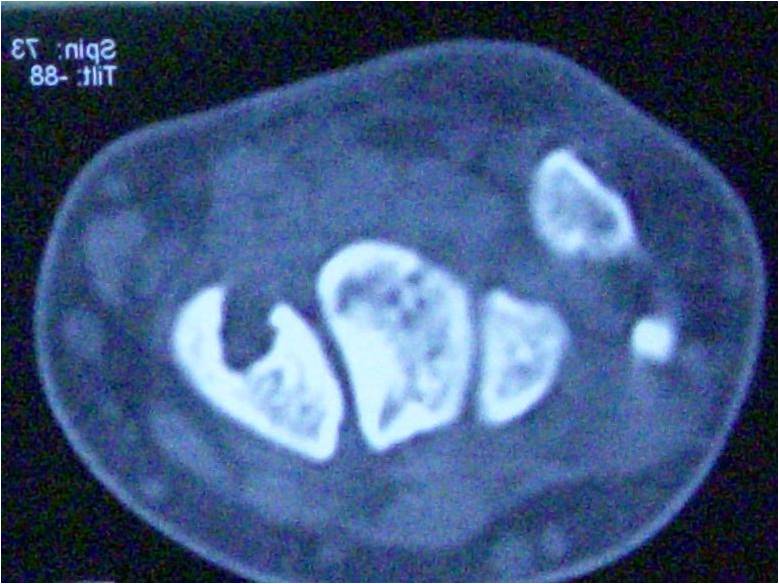
A 4-year-old female presented to genetic OPD for evaluation of short stature. Her weight = 16.2 Kg (median to −2 standard deviation [SD]), height = 92 cm (< −3SD). On examination, the patient had thickened auricular cartilage (Fig. 1) with pre-auricular pits, blue sclerae, dental caries with enamel dysplasia (Fig. 2), syndactyly, brachydactyly, and joint laxity. The patient had unassisted bipedal gait with bilateral intoeing and equinus at the right ankle. There was bilateral genu valgum deformity with patellar subluxation. The clinical tibiofemoral angle was 13° on the right side and 15° on the left side. The Apprehension test for a patellar subluxation was negative. There was an extension lag of 10° on both knees and equinus deformity of 20° at the right ankle. Bilateral metatarsus adductus was present. Radiographs of the pelvis with both hips revealed bilateral rudimentary femoral epiphysis with fragmentation (Fig. 3).
The patient had unassisted bipedal gait with bilateral intoeing and equinus at the right ankle. There was bilateral genu valgum deformity with patellar subluxation. The clinical tibiofemoral angle was 13° on the right side and 15° on the left side. The Apprehension test for a patellar subluxation was negative. There was an extension lag of 10° on both knees and equinus deformity of 20° at the right ankle. Bilateral metatarsus adductus was present. Radiographs of the pelvis with both hips revealed bilateral rudimentary femoral epiphysis with fragmentation (Fig. 3).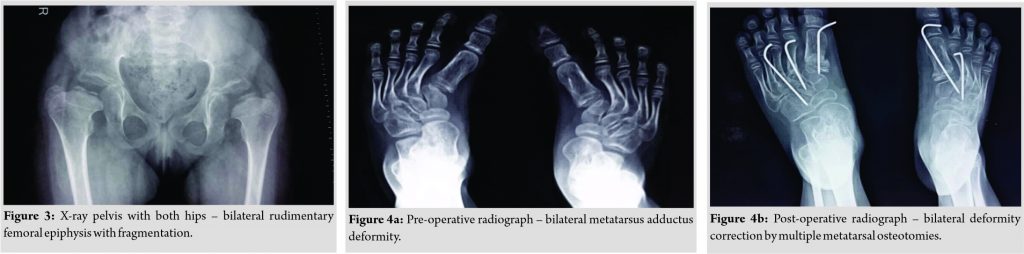
Based on these features, the diagnosis of skeletal dysplasia was suspected, and genotyping was offered to the family after counseling.
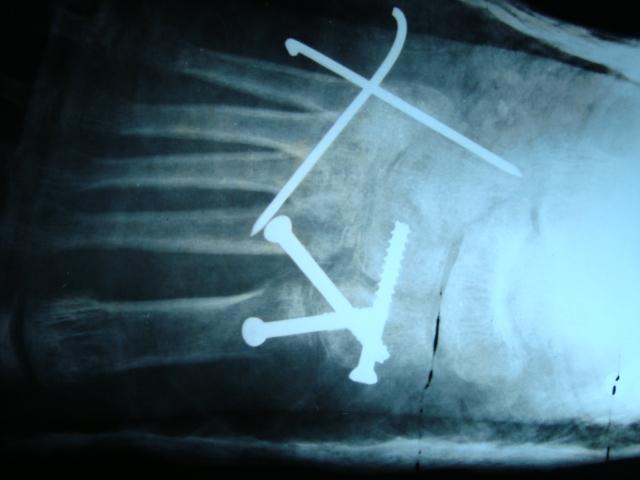
Serial casts for foot deformity were applied but there was no improvement. Metatarsus adductus on each side was corrected by multiple dome osteotomies of metatarsals (Fig. 4a, b). Surgical correction of bilateral genu valgum by medial distal femoral hemiepiphysiodesis was done (Fig. 5a, b). The right side equinus deformity was treated by the Vulpius V-Y plasty of gastrosoleus aponeurosis. On the latest follow-up at 1 ½ years post-surgery, residual genu valgum deformity was noted on the left side and so osteotomy at appropriate age was planned. Clinical correction of metatarsus adductus was achieved with a plantigrade foot (Fig. 6).
Molecular data
Getting the preliminary data and report by NGS, Exon 3 of the SLC26A2 gene was PCR amplified and the product was sequenced using Sanger sequencing. In case of mosaicism in leukocytes, the detection limits of Sanger sequencing for the presence of variation are ~12%. The sequence was aligned to available reference sequence ENST00000286298 to detect variation using variant analysis software programs. (Fig. 7, 8, 9) show variation in the SLC26A2 gene in patient, her father, and mother, respectively.

Discussion
Clinical features of this case bilateral symmetrical cystic ear swelling (hypertrophic ear cartilage), equinus deformity in feet, brachydactyly with ulnar deviation, and short stature are consistent with DTD (due to SLC26A2 mutation) [3]. Persistence of ear abnormalities is seen only in this form of SLC26A2-related disorders (Table 1). The DTDST protein has a complex structure with 12 membrane-spanning domains. Multiple mutations have been noted which result in a myriad combination of clinical features, either through protein truncation or through the reduction in mRNA levels [4]. Mutations leading to amino acid substitutions in the transmembrane portion of protein have severe phenotypical expression than substitutions involving extracellular or cytoplasmic part (N-terminal and C-terminal). It is postulated that there is a leftover activity associated with some mutations and the degree of this leftover activity determines the clinical phenotype [4]. Both skeletal dysplasia and achondroplasia have similar features but morbidity is significantly higher with DTD. Yilmaz et al. in their study consisting of 29 patients of skeletal dysplasia with genu valgum deformity in 38 legs showed hemiepiphysiodesis to be effective in the treatment of genu valgum deformities in skeletally immature patients [5]. Cavovarus deformity which is often present is treated with the Ponseti method of casting in patients in the age group <6 months. There is a need for soft-tissue release in patients with after the walking age and in resistant cases, it may need aggressive release [6]. Metatarsus adductus deformity if resistant may need bony procedures and various ostetomies have been described [7, 8, 9]. A multidisciplinary approach is indicated with the treating team comprising a pediatrician, orthopedic surgeon, audiologist, orthodontists, and physical therapists. Genetic studies have a crucial role in diagnosis, but importance of clinical suspicion (based on certain characteristic deformities) cannot be emphasized more.
Conclusion
Skeletal dysplasias are heterogeneous disorders with widely varying clinical presentations – from mildly symptomatic to overtly fatal. Diagnosis of such cases is tricky in our setup due to lack of investigative set up as well as high costs. However, efforts into a search for diagnosis and dedicated management by a team of skilled clinicians have potential to improve quality of life for patients afflicted by such disorders.
Clinical Message
A multidisciplinary approach is warranted for providing best possible management to such patients. Selected certain anomalies serve as a key feature to identify a disorder (ear abnormalities in this case) and clinicians should be mindful of these. Family counseling regarding prognosis and genetic implication forms the crux of care and helps in improving outcomes, long-term care, and further family planning.
References
1. Krakow D. Skeletal dysplasias. Clin Perinatol 2015;42:301-19.
2. Singh P, Schwarzbauer JE. Fibronectin matrix assembly is essential for cell condensation during chondrogenesis. J Cell Sci 2014;127:4420-8.
3. Bonafé L, Mittaz-Crettol L, Ballhausen D, Superti-Furga A. Diastrophic Dysplasia. Seattle, WA: University of Washington; 2004.
4. Rossi A, Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat 2001;17:159-71.
5. Yilmaz G, Oto M, Thabet AM, Rogers KJ, Anticevic D, Thacker MM, et al. Correction of lower extremity angular deformities in skeletal dysplasia with hemiepiphysiodesis: A preliminary report. J Pediatr Orthop 2014;34:336-45.
6. Al Kaissi A, Kenis V, Melchenko E, Chehida FB, Ganger R, Klaushofer K, et al. Corrections of lower limb deformities in patients with diastrophic dysplasia. Orthop Surg 2014;6:274-9.
7. Steytler JC, Van der Walt ID. Correction of resistant adduction of the forefoot in congenital club-foot and congenital metatarsus varus by metatarsal osteotomy. Br J Surg 1966;53:558-60.
8. Berman A, Gartland JJ. Metatarsal osteotomy for the correction of adduction of the fore part of the foot in children. J Bone Joint Surg Am 1971;53:498-506.
9. Siegel SJ. The modified lepird procedure for correction of metatarsus adductus. J Foot Ankle Surg 2019;58:1045-50.
 |
 |
 |
 |
| Dr. Anchal Kumar Tripathi | Dr. Sunny Choudhary | Dr. Vivek Singh |
Dr. Prashant Kumar Verma |
| How to Cite This Article: Tripathi AK, Choudhary S, Singh V, Verma PK. Genetic Association and Role of Surgery for the Treatment of Lower Limb Deformities in Diastrophic Dysplasia: A Case Report. Journal of Orthopaedic Case Reports 2021 February;11(2): 81-85. |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com