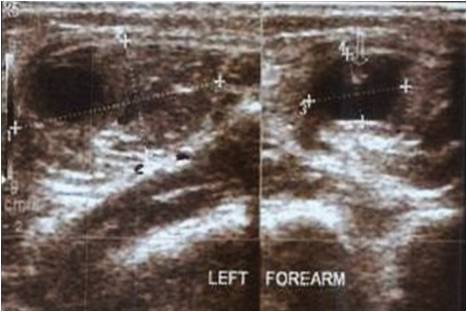
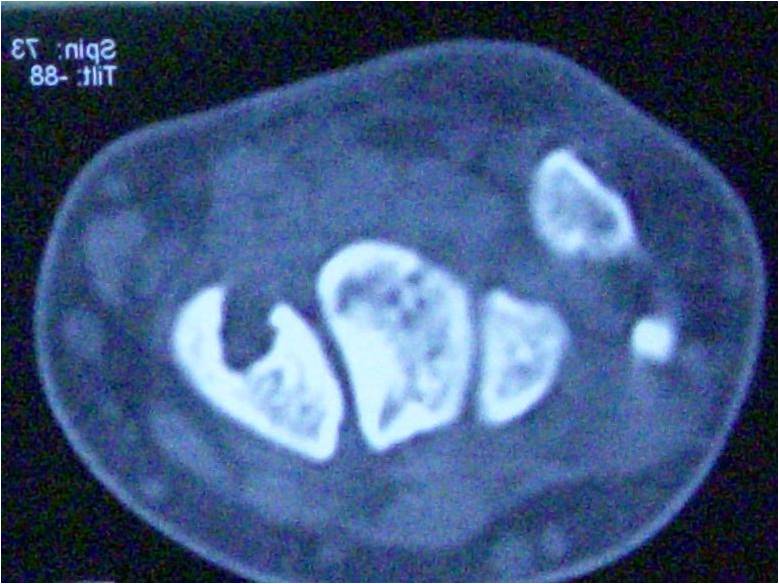
Histopathological confirmation is the key to diagnose different histiocytic disorders.
Dr. Adlyne Reena Asirvatham, Department of Endocrinology, Sri Ramachandra Medical College and Research Institute, Chennai – 600 056, Tamil Nadu, India. E-mail: adlynereena@yahoo.co.in
Introduction:Erdheim-Chester disease (ECD) is a rare non-Langerhans cell histiocytosis (LCH) of unknown origin that was first described in 1930. Since then, almost 600 cases have been reported worldwide. Even though this disease primarily affects the bone, it has a varied clinical spectrum of presentation ranging from asymptomatic bone lesions to multisystem involvement. Owing to its protean manifestations ECD is often misdiagnosed or diagnosed late.
Case Report:We present a 48-year-old female with a long long-standing history of recurrent bone lesion of the tibia and multiple trivial trauma fractures of long bones. Recently, she also developed a persistent headache and painful swelling of the right shoulder and left hip joint. Radiographs revealed multiple lytic and lytic sclerotic lesions. With the probable diagnosis of LCH, she underwent biopsy which revealed features characteristic of ECD.
Conclusion:This case highlights the fact that histopathological confirmation is the key to distinguish various types of histiocytic neoplasms. Overlapping clinical and radiological features with atypical manifestations can occur in both LCH and ECD and does not rule out either of them.
Keywords:Osteolytic lesions, osteosclerotic lesion, histiocytic neoplasm, erdheim chester disease, langerhans cell histiocytosis.
Erdheim-Chester disease (ECD) is a rare non-Langerhans cell histiocytosis (LCH) that was initially described as “Lipoid granulomatosis” by William Chester and Jakob Erdheim in 1930. Since then, almost 600 cases have been reported worldwide [1]. However, the etiology remained unknown until recently when the association of v-raf murine sarcoma viral oncogene homolog B1 (BRAF) mutation in more than 50% of ECD patients has been identified [2]. It primarily affects men between the 5th and 7th decade and is characterized by xanthomatous tissue infiltration with CD68 positive, CD1a negative histiocytes [3]. Even though this disease primarily affects the bone, it has a varied spectrum of presentation ranging from asymptomatic bone lesions to multiorgan involvement. Owing to its protean manifestations ECD is often misdiagnosed or diagnosed late. Patients with ECD typically have symmetric diaphyseal osteosclerotic lesions which is the hallmark finding with or without other system involvement. Osteolytic lesions in ECD are very infrequent and have been reported before [4]. Rarely, isolated lung involvement without bone lesions has also been reported [5]. Here, we report a rare presentation of 48-year-old women diagnosed to have ECD who presented with multiple osteolytic lesions and no systemic manifestations.
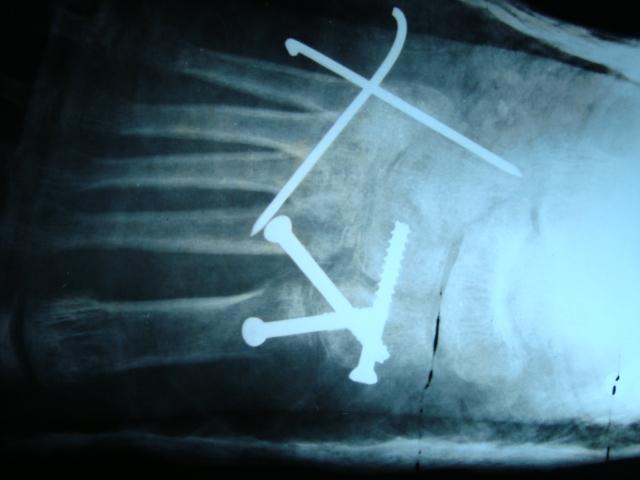
A 48-year-old perimenopausal women presented elsewhere with a 24-year history of difficulty in walking and recurrent bone lesion in the left tibia requiring multiple surgeries. Histopathological examination of the bone lesion was suggestive of multinucleated giant cell tumour and hence underwent local irradiation. She also encountered trivial trauma fractures of the right hip and left femur 4 years and 12 years since the initial symptom onset requiring surgery.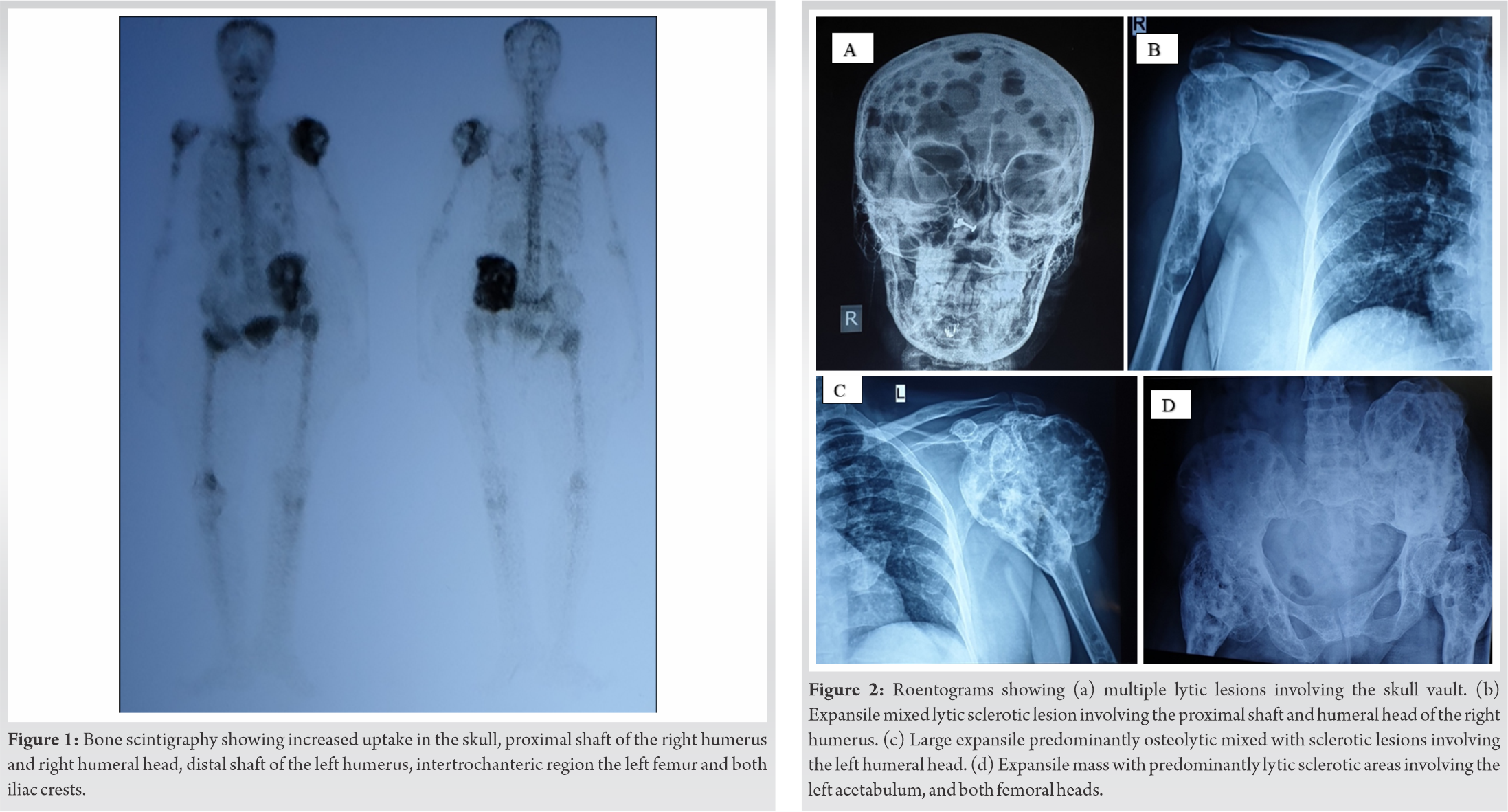 In view of recurrent fractures, she was treated with a single dose of intravenous zoledronate and was lost to follow up. She developed a persistent headache and painful swelling of the right shoulder and left hip joint over the past 5 years and hence sought an Orthopedic consultation and was referred to us to rule out hyperparathyroid bone disease. She had no history to of suggest diabetes insipidus, hypopituitarism, raised intracranial tension, skin, eye, kidney, lung, or cardiac involvement. On physical examination, the patient had no exophthalmos, cutaneous lesions, ataxia, dysarthria, limb weakness, and fundi were normal. Her systemic examination was normal. Local examination showed deformity of the left leg with obvious tender bony swelling of the right shoulder and left hip.
In view of recurrent fractures, she was treated with a single dose of intravenous zoledronate and was lost to follow up. She developed a persistent headache and painful swelling of the right shoulder and left hip joint over the past 5 years and hence sought an Orthopedic consultation and was referred to us to rule out hyperparathyroid bone disease. She had no history to of suggest diabetes insipidus, hypopituitarism, raised intracranial tension, skin, eye, kidney, lung, or cardiac involvement. On physical examination, the patient had no exophthalmos, cutaneous lesions, ataxia, dysarthria, limb weakness, and fundi were normal. Her systemic examination was normal. Local examination showed deformity of the left leg with obvious tender bony swelling of the right shoulder and left hip.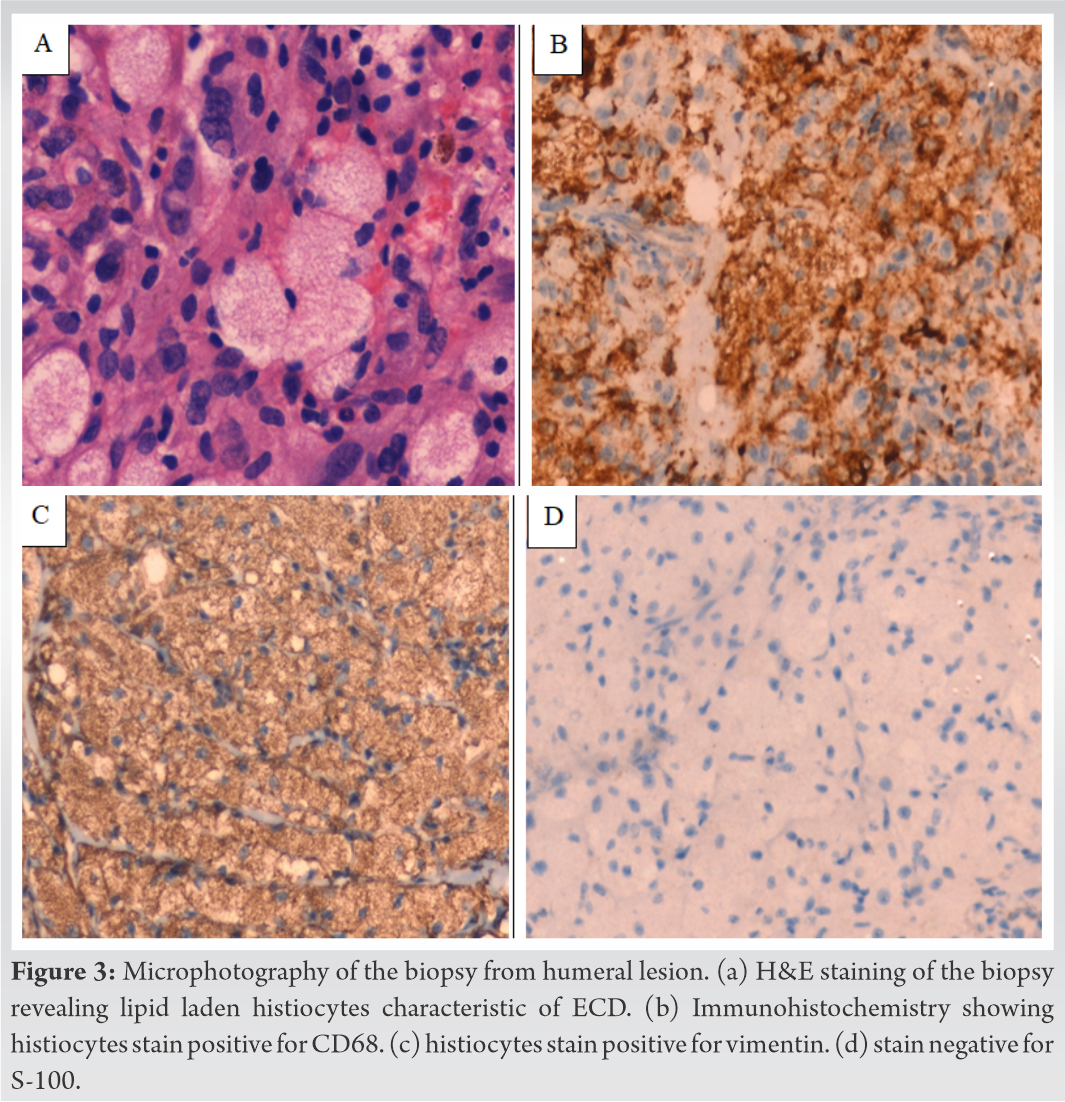
Laboratory analysis showed serum calcium 9.2 mg/dl (8.8-10.6), Albumin 3.2 g/dl (3.5-5.2), Phosphorus 3.6 mg/dl (2.5-4.5), Alkaline phosphatase (ALP) 61 IU/L (32-120), 25-hydroxy vitamin D 22.7 ng/dl (optimal level >30), parathyroid hormone (PTH) 26.3 pg/ml (12-88), serum creatinine 0.6 mg/dl (0.8-1.3). Pituitary functions were normal as shown in (Table 1).
99m Technetium 99m -methyl disphosphonate Tc-MDP Bone Scintigraphy showed discrete hot spots in the skull, mainly in the occipital and parietal region, proximal shaft of the right humerus and right humeral head, distal shaft of the left humerus, intertrochanteric region of the left femur and both iliac crests (Fig. 1). Magnetic resonance imaging (MRI) brain showed no abnormality in the brain parenchyma and pituitary gland but multiple osteolytic lesions in the skull vault and clivus. Computed tomography (CT) chest revealed multiple osteolytic lesions involving the thoracic ribs, clavicle, scapula, and T2 vertebral body. Roentograms showed large mixed expansile predominantly osteolytic with very minimal sclerotic lesions involving the skull, right humerus and left acetabulum, pubic rami, ischium, and both femoral heads (Fig. 2a-2d). Dual-energy X-ray absorptiometry (DXA) scan of the lumbar spine showed normal Bone mineral density BMD (T-score-0.6). Ultrasonogram showed normal kidneys.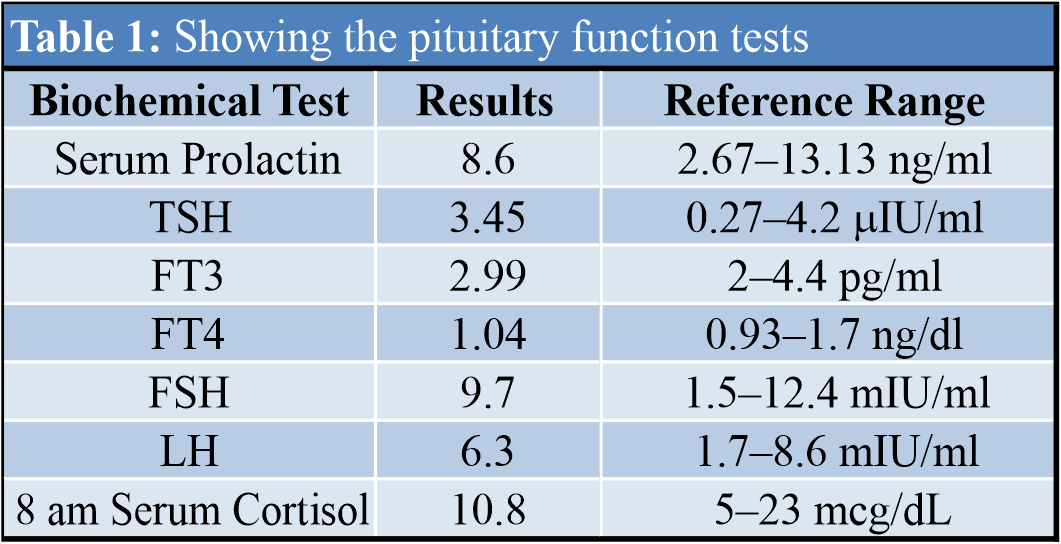
With a probable clinical diagnosis of Langerhans cell histiocytosis (LCH), she was subjected to bone biopsy from the left humerus which showed clusters of small nucleated foamy histiocytes with multinucleated giant cells and mononuclear cells. On immunohistochemistry (IHC), the histiocytes were positive for CD 68, vimentin, and negative for S-100 and CD1a which was characteristic of ECD (Fig. 3a-3d). BRAF mutation analysis was negative and she was advised a therapeutic regimen including interferon-alpha (IFN-a) and chemotherapeutic agents.
Histiocytoses are a group of orphan diseases derived from either macrophages or dendritic cells. Currently, more than 100 subtypes have been identified which are classified 5 different categories namely: L group (Langerhans), C group (Cutaneous and mucocutaneous non-LCHLangerhans cell histiocytosis), M group (Malignant histiocytoses), R group (Rosai-Dorfman disease and miscellaneous non-cutaneous non-Langerhans histiocytoses) and H group (Hemophagocytic lymphohistiocytosis and macrophage activation syndrome [6]. Histiocytoses that affect the skeletal system include Langerhans cell histiocytosis (LCH ) and Erdheim–Chester disease (ECD) both of which are grouped under the Langerhans group since almost 20% of the ECD patients also manifest LCH lesions [6]. Both these disorders are inflammatory myeloid neoplasms caused by mutations in the MEK–extracellular signal-regulated kinase (ERK) signaling pathway, most commonly involving BRAF [7]. While LCH occurs more commonly in children with poor outcome in multisystem disease, ECD occurs predominantly in adults. The onset of symptoms in our case was at 24 years of age which is earlier than the median age reported before. Clinically, ECD is quite different from LCH by the presence of xanthelasma, cardiac (pericardial effusion, coated aorta, and myocardial infiltration), pulmonary (interlobular septal thickening), renal (hairy kidney, and perinephric infiltration), neurological (brain parenchymal and dural-based lesions) involvement [7]. However, our patient had exclusive osseous manifestation of ECD unlike the more common extra-skeleton presentation reported in the literature which could be possibly the reason for misdiagnosis. In addition to the classic clinical features, the pathognomic radiological finding seen in ECD is symmetric diaphyseal and metaphyseal osteosclerosis in the lower limbs which is almost always observed. In our patient, the bony lesions were asymmetric involving the long bones and not sparing craniofacial bones with predominant osteolytic and negligible areas of osteosclerosis alike LCH. This is concurrent with the earlier understanding of both these diseases as a spectrum of hematopoietic disorder possibly explaining the overlap in bone involvement. ECD can present with two different patterns of bone involvement: diffuse skeletal involvement with symmetrical sclerosis typically involving the long bone [8, 9] and the less common pseudotumoral lesions which affect the bone and visceral organs. Two distinct patterns of bone involvement have been observed in ECD: the typical diffuse skeletal involvement, with symmetrical sclerosis of the long bones [8, 9] and focal pseudotumoral lesions involving bone and visceral organs [10]. Only 10% of cases with ECD have been shown to have lytic lesions [9]. Multiple expansile lytic lesions were noted in our case without affecting any other viscera. Less than 10 cases with isolated skeletal lytic lesions have been reported so far with only two presenting with expansile lytic skeletal lesions as our case [11, 12]. Even though LCH and ECD are closely related histiocytic neoplasms, they have distinguishing pathological features to differentiate either of them. Langerhans cells express CD1a, S100 protein and demonstrate the presence of Birbeck granules in electron microscopy whereas in ECD, histiocytes express evidence of phagocytic differentiation with the presence of CD68 immunopositivity and absence of CD1a protein, S100 protein, and Birbeck granules [13]. Haroche et al. 2014 have proposed diagnostic criterion (Table 2) according to which histological confirmation is the gold standard for diagnosing ECD. Despite having atypical clinical and radiological features, the histopathological finding in our case was consistent with ECD thus proving the diagnosis. Recent findings suggest that ECD is a clonal disease and mutational analysis has revealed >50 % mutations in the BRAF proto-oncogene and very rarely in the other genes involved in the MAPK activation pathway [14]. Based on this finding, several small case studies have shown the efficacy of BRAF inhibition (Vemurafenib) in ECD patients harbouring BRAF V600E mutation [13]. Similarly, the involvement of IFN-a and Interleukin (IL-1) have has been well established. These therapeutical options with IFN-a and IL-1 receptor antagonist are recommended as first-line treatment in ECD [15]. In case of non-fatal bone involvement, bisphosphonates may be an ideal option with regards to their cellular properties [16]. Our patient has received Zolendronic acid 1 year back and she is yet to receive IFN-a therapy. Previous studies have reported poor prognosis with the disease but with recent developments in therapy have significantly improved the 5 year survival rate to 68% with therapy [15]. Despite early onset of symptoms, 24-year history, multiple expansile predominant osteolytic lesions, our patient is doing well with moderate quality of life likely due to lack of extraskeletal involvement.
This case highlights the fact that histopathological confirmation is the key to distinguish the various types of histiocytic neoplasms. Overlapping clinical and radiological features with atypical manifestations can occur in both LCH and ECD and does not rule out either of them. The uniqueness of this case is its presentation itself involving only the skeletal system and predominant osteolytic lesions.
Histiocytoses are rare entities derived from dendritic cells or phagocytic/monocytic cells. ECD is characterized by tissue infiltration of CD68 positive, CD1a negative foamy histiocytes. Histopathological confirmation is the key to distinguish the various types of histiocytic neoplasms.
References
- 1.Haroche J, Arnaud L, Cohen-Aubart F, Hervier B, Charlotte F, Emile JF, et al. Erdheim-Chester disease. Curr Rheumatol Rep 2014;16:412. [Google Scholar]
- 2.Cives M, Simone V, Rizzo FM, Dicuonzo F, Lacalamita MC, et al. Erdheim-chester disease: A systematic review. Crit Rev Oncol Hematol 2015;95:1-11. [Google Scholar]
- 3.Bindra J, Lam A, Lamba R, VanNess M, Boutin RD. Erdheim-Chester disease: An unusual presentation of an uncommon disease. Skeletal Radiol 2014;43:835-40. [Google Scholar]
- 4.Krishna VV, James TE, Chang KT, Yen SS. Erdheim-Chester disease with rare radiological features in a 14-year old girl with pre-B acute lymphocytic leukemia and diabetes mellitus. J Radiol Case Rep 2014;8:7-15. [Google Scholar]
- 5.Josan ES, Green JW, Zaidi S, Mehta JB. Isolated pulmonary involvement in Erdheim-Chester disease. Lung India 2017;34:555-8. [Google Scholar]
- 6.Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127:2672-81. [Google Scholar]
- 7.Haroche J, Arnaud L, Cohen-Aubart F, Hervier B, Charlotte F, Emile JF, et al. Erdheim-Chester disease. Rheum Dis Clin North Am 2013;39:299-311. [Google Scholar]
- 8.Pertuiset E, Laredo JD, Lioté F, Wassef M, Jagueux M, Kuntz D. Erdheim-Chester: Description d’un cas, revue de la littérature et discussion des rapports avec les histiocytoses Langerhansiennes [Erdheim-Chester disease: report of a case, review of the literature and discussion of the relation to Langerhans-cell histiocytosis]. Rev Rhum Ed Fr 1993;60:601-9. [Google Scholar]
- 9.Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, Wechsler J, Brun B, Remy M, et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996;75:157-69. [Google Scholar]
- 10.Lyders EM, Kaushik S, Perez-Berenguer J, Henry DA. Aggressive and atypical manifestations of Erdheim-Chester disease. Clin Rheumatol 2003;22:464-6. [Google Scholar]
- 11.Vallonthaiel AG, Mridha AR, Gamanagatti S, Jana M, Sharma MC, Khan SA, et al. Unusual presentation of Erdheim-Chester disease in a child with acute lymphoblastic leukemia World J Radiol 2016;8:757-63. [Google Scholar]
- 12.Singh P, Shrestha R, Yadav NK. Erdheim Chester disease: A subtle quiddity; the first case reported from Nepal. Radiol Case Rep 2020;15:2080-4. [Google Scholar]
- 13.Pineles SL, Liu GT, Acebes X, Arruga J, Nasta S, Glaser R, et al. Presence of Erdheim-Chester disease and Langerhans cell histiocytosis in the same patient: A report of 2 cases. J Neuroophthalmol 2011;31:217-23. Doi:10.1097/WNO.0b013e31820a204e.. [Google Scholar]
- 14.Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121:1495-500. [Google Scholar]
- 15.Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood 2014;124:483-92. [Google Scholar]
- 16.Poiroux L, Paycha F, Polivka M, Ea HK. Efficacy of zoledronic acid in Erdheim-Chester disease: A case report. Joint Bone Spine. 2016;83(5):573-5. [Google Scholar]