At present, no standard of care exists for treatment of patients presenting with MDST, and further identification and evaluation of impacted patients are needed to progressively optimize management..
Dr. Shiraz Younas, Department of Pediatrics, Division of Medical Genetics, McGovern Medical School at the University of Texas Health Science Center at Houston (UT Health Houston) and Children’s Memorial Hermann Hospital, Houston, Texas. E-mail: shiraz.younas@uth.tmc.edu
Introduction: Metaphyseal dysplasia describes a heterogenous group of skeletal dysplasias with varying inheritance patterns, which preferentially demonstrate dysplastic changes within the metaphyseal region of long bones. The clinical consequences of these dysplastic changes are highly variable, but most uniformly include decreased stature, increased upper-to-lower segment proportions, genu varus, and knee pain. Metaphyseal dysplasia, Spahr type (MDST) [MIM: 250400] is a rare primary bone dysplasia that was first clinically described in 1961 in four of five siblings with moderate short stature, metaphyseal dysplasia, mild genu vara, and no biochemical signs of rickets. For many decades, MDST was a clinical diagnosis, but the underlying genetic etiology was determined to be due to biallelic pathogenic variants in matrix metalloproteinases 13 [MIM: 600108] in 2014. Clinical case reports of this disease are limited; this paper aims to present the clinical manifestations and treatment for 3 Filipino siblings with a confirmed of MDST.
Case Report: Patient 1 presented at age 8 for medial ankle pain and bilateral lower extremity bowing of several years. Radiographs showed bilateral metaphyseal irregularities, and the patient underwent bilateral lateral distal femoral and proximal tibial physeal tethering at 9 years 11 months. At 16 months post tethering, she reports reduced pain although varus deformity persists. Patient 2 presented to clinic at age 6 for concern of bilateral bowing. He has had no reported pain and demonstrates milder metaphyseal irregularities than patient 1 on radiographs. To date, patient 2 has no significant changes or gross deformity. Patient 3 examined at 19 months without observable deformity.
Conclusion: Suspicion for MDST should be elevated in the setting of short-stature, upper-to-lower segment disproportionality, focal metaphyseal irregularities, and normal biochemical presentation. At present, no standard of care exists for treatment of patients with these deformities. Further, identification and evaluation of impacted patients are needed to progressively optimize management.
Keywords: Skeletal, metaphyseal, dysplasia, MMP13, MDST.
Metaphyseal dysplasia describes a heterogenous group of skeletal dysplasias with varying inheritance patterns, which preferentially demonstrate dysplastic changes within the metaphyseal region of long bones; however, abnormalities throughout the skeletal system can also be found [1]. The clinical consequences of these dysplastic changes are highly variable, but most uniformly include decreased stature, increased upper-to-lower segment proportions, genu varus, and knee pain [2]. Metaphyseal dysplasia, Spahr type (MDST) [MIM: 250400] is a rare primary bone dysplasia that was first clinically described in 1961 in four of five siblings with moderate short stature, metaphyseal dysplasia, mild genu vara, and no biochemical signs of rickets [3]. Initially, this clinical presentation is highly suggestive of rickets and individuals are often misdiagnosed, but further investigation reveals absence of biochemical abnormalities effectively ruling out rickets as a potential etiology. In addition, MDST lacks extra-skeletal complications unlike rickets [4]. For many decades, MDST was a clinical diagnosis, but the underlying genetic etiology was determined to be due to biallelic pathogenic variants in matrix metalloproteinases (MMP)13 [MIM: 600108] in 2014 [2]. MMP 13 is located at chromosome 11 q22.2, contains 10 exons, and spans approximately 12.5 kb [5]. This gene encodes the MMP 13 protein which is a member of the peptidase M10 family of MMPs. This protein plays a role in the degradation of collagen and other proteins in the extracellular matrix with particular importance in the maturation and remodeling of the cartilaginous component of physes in growing children [2]. Clinical case reports of this disease are limited. This paper aims to present the clinical manifestations and treatment for 3 Filipino siblings with a confirmed diagnosis of MDST.
Here, we report on three siblings born to non-consanguineous parents of Filipino decent. Both mother (160 cm; 25–50th percentile) and father (175 cm; 25th–50th percentile) were of normal height with a predicted mid-parental height of 161 cm for girls (25th–50th percentile) and 174 cm for boys (25–50th percentile). There is no family history of short stature. Of note, there is the presence of three spontaneous miscarriages occurring between Patient 2 and Patient 3. Patient 1, an 8 years and 4 months old female, originally presented to clinic secondary to medial ankle pain when running at school causing her to limp. Her mother noted bowing to the bilateral lower extremity for a few years before this, but it had not yet caused the patient limitations or pain. At this time, she was 118.5 cm (3rd percentile; 50th percentile for an ~6.5-year-old) in height and 23.6 kg (22nd percentile). Patient 1 was the first-born child through spontaneous vaginal delivery at 36 weeks with no complications. She was diagnosed with jaundice at the time of birth but required no intervention and this spontaneously resolved. Growth parameters at birth, per parental recall, were a weight of 2.38 kg (10–25th percentile) and a length of 43 cm (3–10th percentile). She met all developmental milestones appropriately throughout childhood.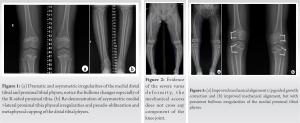
Physical examination at the time of clinic presentation demonstrated symmetric facies with no blue discoloration of her sclerae. She had no asymmetry nor abnormality to hair, teeth, or nails. Upper extremities were symmetric and proportional to head and torso. She was short in stature with gross genu varum alignment through the bilateral lower extremities and her lower segment appeared disproportionate to her upper segment. She maintained full range-of-motion (ROM) through bilateral hips, knees, and ankles and was non-tender to palpation throughout. Radiographs demonstrated bilateral metaphyseal irregularities and asymmetry within the distal femoral physes and proximal tibia physes. Asymmetric focal bulbous appearance of the medial proximal tibial physes compared to the lateral aspect was present bilaterally (Fig. 1a). Bilateral distal tibia demonstrated physeal cupping with asymmetric irregularities both medially and laterally (Fig. 1b). Long-leg standing films were obtained and demonstrated a mechanical axis propagating through the medial compartment bilaterally, right greater than left. Measurement of the patient’s alignment demonstrated a mechanical lateral distal femoral angle (mLDFA) of 87.6° and 89°, medial proximal tibial angle (MPTA) of 85.7° and 81.4°, and lateral distal tibial angle (LDTA) of 91.8° and 93.7° to the right and left lower extremities, respectively.
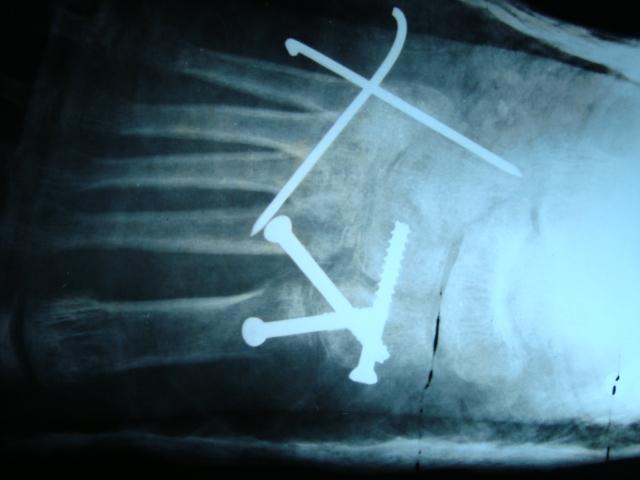
The patient was advised to begin at-home therapy exercises including stretching and strengthening and was sent for laboratory evaluation of Vitamin D, Calcium, Magnesium, and Phosphorus. Each of these laboratory results were within normal limits. The patient re-presented at age 9 years and 11 months with significant progression of genu varum and increased pain at the ankles bilaterally. She described intermittent pain in the medial knees as well. At that time, she was 124 cm (1st percentile; 50th percentile for an ~7.5 years old) in height and 30 kg (35th percentile) in weight. Gross evaluation of the limbs demonstrated marked progression of her varus deformity. Repeat radiographs demonstrated persistent metaphyseal changes and progression of her mechanical axis to a location medial to the medial compartment of the knees bilaterally (Fig. 2). Alignment radiographs demonstrated mLDFA of 92.8° and 90.6°, MPTA of 80.8° and 76.8°, and MDTA of 94.5° and 96.7° to the right and left lower extremities, respectively. Discussion was had with the patient and her family, and ultimately, the patient underwent bilateral lateral distal femoral and proximal tibial physeal tethering. She tolerated the procedure well with no complications. Subsequently, this patient was evaluated by genetics and a skeletal dysplasia panel was performed at a commercial laboratory. This panel performed sequencing and deletion duplication analysis on 109 genes and identified a pathogenic variant in MMP13 [NM002427.3], c.619T > G (p.Trp207Gly) and a variant of uncertain significance (VUS) in MMP13, and c.539G > C (p.Gly180Ala). Due to the proximity of these changes, the laboratory was able to determine these variants that were in trans. At her most recent clinic visit, the patient is 16 months status post tethering procedure and doing well. Gross varus deformity persists; however, pain in the ankles and knees has improved. Repeat radiographs demonstrate minimal change in mechanical axis – now passing through the very medial aspect of the medial compartment bilaterally (Fig. 3). Focal angular measurements have improved in the proximal tibia bilaterally but continue to progress in both the distal femur and distal tibia. Alignment radiographs demonstrate mLDFA of 97° and 93.1°, MPTA of 87.1° and 82.5°, and MDTA of 101.3° and 92.7° to the right and left lower extremities, respectively. Patient 2 was the second born child through induced vaginal delivery at 36 weeks secondary to intrauterine growth retardation (IUGR). On delivery, the patient had mild jaundice which resolved spontaneously; he had no further complications or medical issues. Growth parameters at birth, per parental recall, were a weight of 2.3 kg (25th percentile) and a length of 45.7 cm (10–25th percentile). He met all developmental milestones throughout early childhood. He presented to clinic at age 6 years, 1 month with concern for bilateral bowing of the lower extremities. At time of presentation, he was 95 cm (4th percentile) in height and 14 kg (9th percentile) in weight. Unlike his sister, patient 2 reported no pain or limitations in activities. On examination, he had symmetric facies with no blue discoloration of his sclerae. He had no asymmetry or abnormality to hair, teeth, or nails. Upper extremities were symmetric and proportional to head and torso. He was short in stature with disproportionate upper-to-lower segment distribution and an abnormal circumduction gait. Gross genu varum deformity was mild, but evident. He maintained full ROM to bilateral hips, knees, and ankles and was non-tender to palpation throughout. Radiographs demonstrated mild irregularities to the metaphyseal region of the bilateral distal femoral, proximal tibial, and distal tibial physes; these were less severe than those seen in the patient’s older sister. Overall alignment demonstrated mild varus, left worse than right, with a mechanical axis through the central portion of the medial compartments bilaterally (Fig. 4). Alignment radiographs demonstrated a mLDFA of 88.4° and 97.2°, MPTA of 85.1° and 88.1°, and LDTA of 97° and 92° to the right and left lower extremities, respectively. Patient 2 was similarly evaluated by genetics with his older sister; the same skeletal dysplasia panel was performed and demonstrated identical genetic variants as his sister (above). He was monitored intermittently over the course of the next 2 years with no significant change in gross deformity. He reported occasional bilateral medial knee pain that resolved spontaneously in all cases. At 8 years and 7 months, the patient remains short-statured with a height of 121 cm (3rd percentile, 50th percentile for a 7 years old). Repeat alignment radiographs demonstrate a mLDFA of 91.2° and 94.5°, MPTA of 86.7° and 88.0°, and LDTA of 94° and 91.5° to the right and left lower extremities, respectively. Discussion has been had about potential physeal tethering in the future, but, at this point, the family is content with maintained observation. Patient 3 was the third born, but 6th pregnancy, to the same non-consanguineous parents through induced vaginal delivery at 36 weeks secondary to IUGR. On delivery, she had no further complications or medical issues. Growth parameters at birth were a weight of 2.384 kg (10–25th percentile) and length of 47 cm (10–25th percentile). She has met all developmental milestones up to time of presentation. She presented to clinic with her mother at 1 year and 7 months of age due to “popping” of her shoulder joints and concern for bilateral lower extremity bowing. She was 76.4 cm (5–10th percentile) in length and 8.9 kg (10th percentile) in weight. On examination, the patient has symmetric facies with no blue discoloration of her sclerae. She has no abnormality to hair, teeth, or nails. Upper extremities are symmetric with full ROM; no “popping” was able to be elicited on examination. No observable gross deformities to the bilateral lower extremities were identified. Due to the family history, the patient had targeted testing for the variants identified in her siblings and was also found to have the same variants, consistent with a diagnosis of MDST. She will continue to be monitored as she develops over the course of her childhood.
It should be noted that our sib-ship has followed a clinical course consistent with previously reported individuals with MDST. They were normally grown at birth, despite concerns of IUGR in patient 2 and 3 during pregnancy. Patient and 1 and 2 demonstrated progressive disease with decreasing height centiles after the newborn period. Finally, the presence of pain can be variable depending on severity and is reported to resolve with age. Finally, skeletal involvement is limited to the tibia and distal femur. Previously, nearly all patients were treated with observation, except for one of two patients presented by Tadros et al. [2] who was treated with physeal tethering at the knees bilaterally due to persistent pain and deformity. The patients presented within this case report are similar in clinical presentation to previously reported cases; however, differences do exist. Similar to those reported by Li et al. [11] and Balasubramaniyan et al., [4] our patients’ parents were non-consanguineous – a characteristic that varies from classic presentation of those diagnosed with MDST. Our patient 1 demonstrated progressive deformity and increasing pain over the course of her clinical management, which ultimately prompted surgical intervention through the use of bilateral distal femoral and proximal tibial physeal tethering. Unfortunately, while some correction was obtained in the MPTA, deformity at the mLDFA and LDTA continued to progress despite the surgical intervention. Surprisingly, however, this progression did not correlate with persistent pain, as our patient’s ankle and knee pain resolved. This finding may support an approach of continued observation in future patients even in the setting of progressive deformity due to the ineffectiveness of surgical intervention with attempts at growth modulation through tethering. More definitive angular correction through osteotomy may be necessary at an older age once the patient no longer demonstrates progressive deformity and physes have closed. Observation has been employed as the primary intervention currently for patient 2 and patient 3 in our case report.
The 2019 revised Nosological Classification of Skeletal Disorders group metaphyseal dysplasias as a set of disorders that are hallmarked by radiographic evidence of metaphyseal irregularities and asymmetries in the long bones [6]. Despite this commonality, the overall severity and phenotypic spectrum of this nosologic group (Group 11) is broad including variations in age of onset, extent of skeletal involvement, and extra-skeletal manifestations. These diseases commonly manifest with short stature, disproportionate upper-to-lower segment ratios, altered gait, genu varum, and knee pain. Pathogenic variants leading to these disorders are frequently located in genes responsible for factors involved in cartilaginous remodeling and growth. To date, pathogenic variants in 11 different genes leading to ten distinct metaphyseal dysplasias have been identified demonstrating both autosomal dominant and autosomal recessive inheritance as well as pleiotropy and locus heterogeneity. Historically, metaphyseal anadysplasia (MAD) type 1 was thought to be inherited through autosomal dominant and autosomal recessive patterns through pathogenic variants in MMP13. In Lausch et al. 2009, there was one individual (family D) that was reported to have homozygous pathogenic variants (c.722C > A, p.H213N) in MMP13 and at the time was given a diagnosis of autosomal recessive MAD type 1 [10]. Bonafe et al., in 2014, reviewed that family and determined that individual did not have MAD type 1, but rather a diagnosis of MDST [1]. Their data suggests that autosomal dominant MAD type 1 is caused by gain of function variants in MMP13. Contrarily, MDST is caused by biallelic loss of function variants in MMP1, specifically the gain of function variants in the catalytic domain between amino acids 172 and 232 of the MMP13 protein. As a result, the gain of function variants leads to heterocatalytic self-inactivation and produces a dominant negative effect. Similarly, MDST biallelic pathogenic variants are commonly due to loss of function variants in the catalytic domain. The underlying genetic etiology of these two conditions supports the similar clinical presentation of autosomal dominant MAD type 1 and MDST. This information supports the retrospective diagnosis of MDST in the individual from family D as well as our patients. The pathogenic variant and VUS identified in our patients are in the catalytic domain. Notably, the commercial laboratory reported that the VUS is suggestive of loss of function and is observed in individuals with clinical features of autosomal recessive metaphyseal dysplasia. In addition, the pathogenic variant identified in this family has been previously reported in other individuals with MDST as well. Therefore, we are confident that MDST is the diagnosis for this sibling set. While the generalized radiographic presentation of these disorders is similar, there are small differences that distinguish each individual diagnosis. There is importance in distinguishing between the individual diagnoses as the prognostic implications are different [1]. Since the initial report describing patients with characteristics of MDST, eight additional case reports have been published to our knowledge [1-4, 8-11]. Excluding two case reports [4,11], all cases were reported to have occurred in consanguineous relationships. Clinical presentation has been similar throughout, with most patients presenting within the first decade of life with short-stature and genu varum of the bilateral lower extremities. The consistent difference between MAD type 1 and MDST is regarding stature. Individuals with MAD are typically short statured during childhood but have adult height between -1 and -2 standard deviation from the mean. In contrast, individuals with MDST have short stature in childhood and adulthood. Comparing MAD type 1 and MDST to other metaphyseal dysplasias, there are unique and differentiating features including extra-skeletal abnormalities and spinal abnormalities that are not present in MAD type 1 or MDST. This emphasizes the importance of detailed radiographic evaluation, family history, and molecular testing for all patients with clinical features suggestive of metaphyseal dysplasia. Differentiation of these different diagnoses is also important for appropriate recurrence risk cancelling in patients and families.
MDST is a rare disease occurring in <1 in 1,000,000 live births [12]. Frequently, patients with MDST are initially misdiagnosed with rickets due to the similar appearance of lower extremity bowing and genu varum. However, suspicion for MDST should be elevated in the setting of short-stature, upper-to-lower segment disproportionality, focal metaphyseal irregularities, and normal biochemical laboratory values – all symptoms inconsistent with a diagnosis of rickets. Genetic consultation and further testing should be encouraged to obtain a proper diagnosis. At present, no standard of care exists for treatment of patients with these deformities. However, our experience with relatively unsuccessful correction of alignment through bilateral distal femoral and proximal tibial physeal tethering may represent an ineffectiveness of growth modulation and a preference for observation in these patients despite progressive deformity. Further, identification and evaluation of patients with this disease are needed to optimize management moving forward.
In the coming years, the rising prevalence of genetic testing will result in increased diagnosis of rare skeletal dysplasia’s such as MDST. No standard of care currently exists for MDST, and further, identification/evaluation of afflicted patients will enable optimization of management.
References
- 1.Bonafé L, Liang J, Gorna MW, Zhang Q, Ha-Vinh R, Campos-Xavier AB, et al. MMP13 mutations are the cause of recessive metaphyseal dysplasia, Spahr type. Am J Med Genet A 2014;164A:1175-9. [Google Scholar]
- 2.Tadros S, Scott RH, Calder AD, Hurst JA. Metaphyseal dysplasia, Spahr type; Missense MMP13 mutations in two Iraqi siblings. Clin Dysmorphol 2017;26:13-7. [Google Scholar]
- 3.Spahr A, Spahr-Hartmann I. Familial metaphysial dysostosis: Study of 4 cases in siblings. Helv Paediatr Acta 1961;16:836-49. [Google Scholar]
- 4.Balasubramaniyan M, Kaur A, Sinha A, Gopinathan NR. Metaphyseal dysplasia, Spahr type: A mimicker of rickets. BMJ Case Rep 2019;12:e230257. [Google Scholar]
- 5.Pendas AM, Balbin M, Llano E, Jimenez MG, Lopez-Otin C. Structural analysis and promoter characterization of the human collagenase-3 gene (MMP13). Genomics 1997;40:222-33. [Google Scholar]
- 6.Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A 2019;179:2393-19. [Google Scholar]
- 7.Lausch E, Keppler R, Hilbert K, Cormier-Daire V, Nikkel S, Nishimura G, Unger S, Spranger J, Superti-Furga A, Zabel B. Mutations in MMP9 and MMP13 determine the mode of inheritance and the clinical spectrum of metaphyseal anadysplasia. Am J Hum Genet 2009;85:168-178. [Google Scholar]
- 8.Farag TI, Teebi AS. The second family with Spahr-type metaphyseal chondrodysplasia: Autosomal recessive inheritance confirmed. Clin Genet 1990;38:237-9. [Google Scholar]
- 9.Mégarbané A, Chouery E, Ghanem I. A multiplex family with possible metaphyseal Spahr-type dysplasia and exclusion of RMRP and COL10A1 as candidate genes. Am J Med Genet A 2008;146A:1865-70. [Google Scholar]
- 10.Lausch E, Keppler R, Hilbert K, Cormier-Daire V, Nikkel S, Nishimura G, et al. Mutations in MMP9 and MMP13 determine the mode of inheritance and the clinical spectrum of metaphyseal anadysplasia. Am J Hum Genet 2009;85:168-78. [Google Scholar]
- 11.Li D, Weber DR, Deardorff MA, Hakonarson H, Levine MA. Exome sequencing reveals a nonsense mutation in MMP13 as a new cause of autosomal recessive metaphyseal anadysplasia. Eur J Hum Genet 2015;23:264-6. [Google Scholar]
- 12.Metaphyseal Chondrodysplasia, Spahr Type. Orphanet Encyclopedia; 2020. Available from: https://www.orpha.net /consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=2501. Accessed 10 July 2022. [Google Scholar]