Fibrodysplasia Ossificans Progressiva, Rare condition, its clinical presentation, management, and outcome
Dr. Nitin Rawal, Department of Orthopaedics, SGT Medical College Hospital and Research Institute, Gurugram, Haryana, India. E-mail: nitin.nini@gmail.com
Introduction: It is an autosomal dominant genetic disease presented with heterotopic ossification of connective tissues after birth and a defect of the big toes. One in ten million births is affected by it worldwide. As a result, diagnosis and management of fibrodysplasia ossificans progressiva (FOP) can be delayed or misdiagnosed. Clinical assessment, radiographic examination, and genetic study of the Activin receptor Type 1A gene are among the diagnostic techniques used to identify this disease.
Case Report: We are presenting three female cases having FOP in this article of different age groups. It presented with multiple non-tender lumps on patients’ paravertebral region along with bilateral hallux valgus. The radiograph revealed ossifications of soft tissue involving spine and neck. The patient was given a conservative treatment approach and told what could be done to prevent flare-ups.
Conclusion: Being a rare, progressive, and often misdiagnosed condition, early diagnosis is advocated. Long-term physiotherapy and muscle trauma prevention can delay it as much as possible to prevent future disabilities.
Keywords: ACVR1 gene, dystrophic ossification, hard bony nodule, heterotopic ossification, hypoplastic great toe, myositis ossificans progressive, ossifications of soft tissue.
Myositis ossificans progressiva is a rare autosomal dominant connective tissue disorder known as fibrodysplasia ossificans progressiva (FOP). The process of heterotopic ossificans and distinct skeletal deformities is hallmarks of the condition. A disfiguration occurs, as well as an inhibition of normal motor function due to the formation of osseous masses that create bridges between sections of the skeleton [1, 2]. A sporadic disease without a geographical or racial origin, it is prevalent throughout the world. One in ten million people has the disease. Patients are often in their first two decades of life and have an average age of 28.7 [3, 4]. A recurrent heterozygous activating genetic mutation leads to FOP, which is associated with bone morphogenic protein (BMP) Type 1 signaling endochondral ossification through the intracellular glycine-serine rich domain of Activin receptor type 1A/activin-like kinase 2 [ACVR1/ALK2]. FOP patients are induced to produce extra skeletal bone through activation of the BMP Smad 1/5/8 signaling tract [3, 5, 6]. We are reporting three cases who presented with multiple hard nodules on the back region. The most probable diagnosis was FOP based on the clinical and radiographic examination. All investigations were conducted in accordance with the institution’s ethical principles. Before participating in the study, the patient provided informed consent.
Case
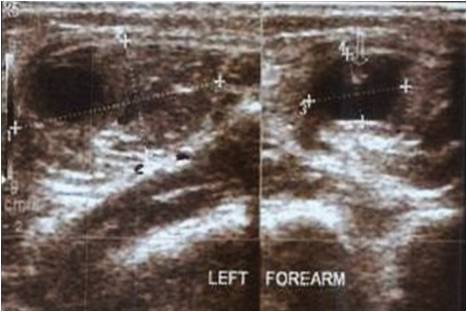
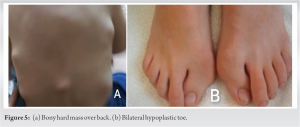
A 12-year-old patient came to our hospital, she had been experiencing back pain and non-tender masses for 6 months. The mass was first noticed when the patient had a fall from the bed 15 months back in occipitocervical region. Since then, the masses grew both in number and size. The patient’s physical examination revealed no abnormalities in her organs or general condition. The paravertebral area, from the cervical region to the lumbar region and scapular region, had lumps ranging in size from 20 to 30 mm (Fig. 1).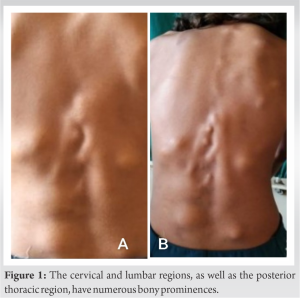 The masses were non tender and no local signs of inflammation were present. The big toes were shortened, and bilateral hallux valgus was present. There was no medical history in the patient’s family. The birth was uneventful and by full-term vaginal delivery.
The masses were non tender and no local signs of inflammation were present. The big toes were shortened, and bilateral hallux valgus was present. There was no medical history in the patient’s family. The birth was uneventful and by full-term vaginal delivery.
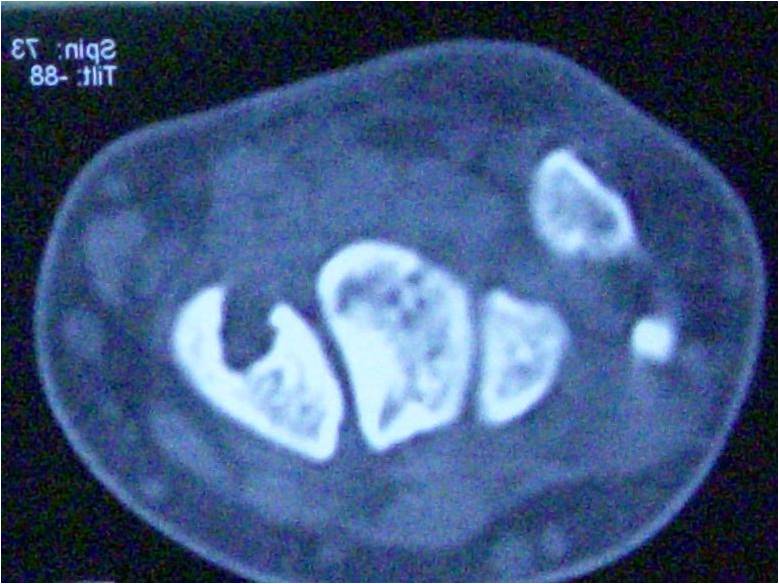
The laboratory studies’ results were standard. HLA B27 was negative. A computed tomography (CT) and magnetic resonance imaging were performed in several areas related to the mass. The radiograph revealed ossifications of soft tissue involving spine and neck (Fig. 2). The CT scan revealed subcutaneous soft-tissue ossification on both sides of T8-L5 paravertebral region, dystrophic ossification of fascia, and intermuscular region between semispinalis capitis muscle. The dystrophic ossification partly formed a bony bridge from fascia and intramuscular part of bilateral levator scapulae muscle, spinalis cervicis muscle, splenius capitis muscle, trapezoid muscle, and rhomboid muscle until erector spinae muscle. Bony destruction was not present, and the spine’s alignment and curvature were normal.
We identified the patient as having FOP based on their clinical and radiological symptoms without doing an unneeded biopsy. The patient’s condition is closely monitored, and NSAIDS drugs have been prescribed for a short period of time, as well as physical therapy to improve everyday activities and cardiopulmonary efficiency. We had informed the parents regarding the nature and progression of the disease. The patient has been on regular follow-up and care was taken, such as biopsy or surgery, or IM injections, to prevent a flare-up.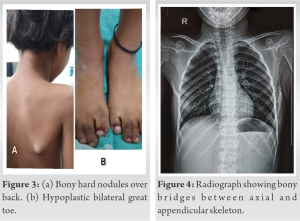
Case 2
A 9-year-old female child presented with the complain of gradual development stiffness of neck and hard bony nodule over the upper back. On examination stiffness in neck, limitation of the left shoulder movements found. She also had a hypoplastic first toe and her left scapula protruded (Fig. 3). She had ectopic ossificans in the paravertebral muscles (Fig. 4). It was difficult for her to open mouth, swallow food, and perform thoracic inspiration. Oral or intravenous administration of ascorbic acid and bisphosphonate was prescribed for the patient significantly improved patient’s quality of life. Being a rare condition, treatment guideline is not clear and need further research.
Case 3
An 8-year-old female with the complain of gradual development hard bony nodule over the upper back. She also had hypoplastic great toe and her scapula was protruded. Other Radiological findings were lost in follow-up. She was prescribed NSAID’s and ascorbic acid for improvement in functional status. No surgery or biopsy was done due to risk of flaring up of disease.
The first case of FOP was reported in 1692 by Guy Patin in a boy [7]. FOP, an autosomal dominant condition marked by heterotopic ossification, is extremely uncommon.. Progressive ossificans is a disorder that is defined by congenital malformations of the first toes; it typically affects the neck, chest, and back [8, 9]. Bilateral hallux valgus and hard tissue swellings over the afflicted muscles are symptoms of FOP in its early stages. Unprompted or trauma-related flare-ups are typical from birth until the second decade of life [10]. Injury, surgical procedures, overexertion, influenza, and vaccinations can precipitate the formation of lesions. Inflammatory cytokines and migratory factors are released following soft-tissue injury, predominantly in muscle, causing FOP lesions.11 Smad1, Smad5, and BMP-412 are all potential mediators. PGE 2 anabolic qualities might improve osteoblast differentiation in vitro by encouraging the incorporation of non-adherent mesenchymal cells into pre-osteoblasts on bone marrow stromal cells [11]. Plasma levels of PG-like molecules are elevated in patients with FOP. These observations suggest that circulating osteogenic cells may be recruited to facilitate extraskeletal ossification in FOP. At birth, the only congenital defect of the first toes seen in FOP children [13]. Heterotopic ossification normally starts in the paraspinal muscles of the cervical spine during the first decade of life and progresses from axial to appendicular, cranial to caudal, and proximal to distal regions, as in the present case. In axial and appendicular skeletons, heterotopic ossification gradually resulted in ankylosis of all major joints. Eventually bridges are formed between axial bones through the ossification process [14]. By their second decade, the majority of FOP sufferers are restricted to a wheelchair or couch. Cardiopulmonary failure is a frequent reason for death in FOP patients [15]. The three basic criteria for diagnosing FOP are congenital defect of the first toes, progressive heterotopic ossification, and a clearly defined anatomical and temporal pattern. ESR may slightly rise when a flare-up occurs. ACVR 1 gene mutation test is affirmative test. The heterotopic bones are visible in radiographs and CT examinations. FOP should be distinguished from other conditions with symptoms that resemble it, such as progressive osseous heteroplasia, Albright hereditary osteodystrophy, cutaneous ossification, Bechterew disease, systemic juvenile idiopathic arthritis, Klippel-Feil syndrome, short digits, Juvenile hallux valgus, and mesenchymal neoplasms [9]. FOP does not currently have active treatment. Ectopic ossification should be avoided by avoiding potential trigger conditions. Severe pain, limitation of joints, or compression of the nerves may require surgical excision. Myositis ossificans is usually treated surgically when it is ripe. Ossification can be triggered by surgical therapy aimed at improving mobility [3]. In patients with FOP, medications with a theoretical application like leukotriene inhibitors and recent drugs like signal transduction inhibitors and monoclonal antibodies targeting ACVR 1 may show favorable results. Molecular research in FOP has reached the genetic level, but not yet to the point of producing a gene therapy [16]. Physical therapy should concentrate on improving daily activities by avoiding passive ROM that can cause disease flare-ups. Most recent therapy recommendations written by Kaplan et al. followed for our patient management [17]. As soon as flare-ups begin, corticosteroids are recommended. A flare-up that involves the back, neck, or trunk should not be treated with corticosteroids because of their long duration, recurrent nature, and unpredictable onset. An NSAID or a COX-2 inhibitor can be utilized symptomatically when prednisone is withdrawn (in combination with leukotriene inhibitor) [17]. In this case, we avoided corticosteroids as the initial flare-up was already over and patient needed symptomatic treatment for pain. Availability and cost of leukotriene were an issue for starting this drug.
As a rare disease, FOP is often misdiagnosed, resulting in a late diagnosis. Diagnoses and treatments are more likely to be successful if they are performed at an early stage. It is difficult to stop this disease due to its progressive nature, but long-term physiotherapy and muscle trauma prevention can delay it as much as possible to prevent future disabilities. The future research in molecular genetics will help us comprehend the pathophysiology of FOP and develop effective treatments.
By long-term physiotherapy and muscle trauma prevention, the future disabilities can be prevented.
References
- 1.Connor JM, Evans DA. Fibrodysplasia ossificans progressiva: The clinical features and natural history of 34 patients. J Bone Joint Surg 1982;64:76-83. [Google Scholar]
- 2.Hagiwara H, Aida N, Machida J, Fujita K, Okuzumi S, Nishimura G. Contrast enhanced MRI of an early preosseous lesion of fibrodysplasia ossificans progressiva in a 21-month-old boy. Am J Roentgenol 2003;181:1145-7. [Google Scholar]
- 3.Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 2005;116:e654-61. [Google Scholar]
- 4.Thickman D, Bonakdar-pour A, Clancy M, Van Orden J, Steel H. Fibrodysplasia ossificans progressiva. Am J Roentgenol 1982;139:935-41. [Google Scholar]
- 5.Kaplan FS, Seeman P, Connor DL, Glaser L, Carroll P. Classical and atypical fibrodysplasia ossificans progressiva phenotypes are caused by mutations in bone morphogenic proteins Type 1 receptor ACVR1. Hum Mutat 2009;30:379-90. [Google Scholar]
- 6.Pang J, Zuo Y, Chen Y, Song L, Zhu Q, Cai Z, et al. ACVR1-FC suppresses BMP signaling and chondro-osseous differentiation in an in vitro model of fibrodysplasia ossificans progressiva. Bone 2016;92:29-36. [Google Scholar]
- 7.Goldman AB. Heritable diseases of connective tissue, epithelial dysplasia, and related conditions. In: Diagnosis of Bone and Joint Disorders. 4th ed. Philadelphia (PA): W.B. Saunders; 2002. p. 4409-15. [Google Scholar]
- 8.Wei J, Jia Y, Liang B. Myositis ossificans of the serratus anterior as a rare complication of massage: A case report. J Med Case Rep 2015;9:143. [Google Scholar]
- 9.Kaplan FS, Xu M, Glaser DL, Collins F, Connor M, Kitterman J, et al. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics 2008;121:e1295-300. [Google Scholar]
- 10.Feldman G, Li M, Martin S, Urbanek M, Urtizberea JA, Fardeau M, et al. Fibrodysplasia ossificans progressiva, a heritable disorder of severe heterotopic ossification, maps to human chromosome 4q27-31. Am J Hum Genet 2000;66:128-35. [Google Scholar]
- 11.Scutt A, Bertram P. Bone marrow cells are targets for the anabolic actions of prostaglandin E2 on bone: Induction of a transition from nonadherent to adherent osteoblastic precursors. J Bone Miner Res 1995;10:474-87. [Google Scholar]
- 12.Levitz CL, Cohen RB, Zasloff MA. The role of prostaglandins in the pathogenesis of fibrodysplasia ossificans progressiva. Calcif Tissue Int 1992;50:387. [Google Scholar]
- 13.Nakashima Y, Haga N, Kitoh H, Kamizono J, Katagiri T, Tozawa K, et al. Deformity of the great toe in fibrodysplasia ossificans progressiva. J OrthopSci 2010;15:804-9. [Google Scholar]
- 14.Mahboubi S, Glaser DL, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva. Pediatr Radiol 2001;3:307-14. [Google Scholar]
- 15.Kaplan FS, Zasloff MA, Kitterman JA, Shore EM, Hong CC, Rocke DM. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am 2010;92:686-91. [Google Scholar]
- 16.Kaplan FS, Shore EM, Pignolo RJ. The medical management of with fibrodysplasia ossificans progressive: Treatment considerations. Clin Proc Int Clin Consort FOP 2011;4:1-100. [Google Scholar]
- 17.Kaplan FS, Al Mukaddam M, Baujat G, Brown M, Cali A, Cho TJ, et al. The medical management of with fibrodysplasia ossificans progressive: Current treatment considerations. Proc Int Clin Council FOP 2019;1:1-111. [Google Scholar]