
The article emphasizes the diagnostic challenges and critical role of histopathological evaluation in accurately diagnosing rare lymphoproliferative neoplasms, such as Rosai-Dorfman disease, Kimura disease, and Castleman disease, which often present with atypical musculoskeletal manifestations and are frequently misdiagnosed as non-neoplastic conditions.
Dr. Meagan E Tibbo, Department of Orthopedic Surgery, Mayo Clinic, 5777 E Mayo Blvd, Phoenix 85054, AZ, USA. E-mail: tibbo.meagan@mayo.edu
Introduction: Lymphoproliferative neoplasms (LPNs) can lead to diverse musculoskeletal manifestations, presenting diagnostic challenges due to their rarity and atypical clinical features.
Case Report: This retrospective case series, conducted at a tertiary care hospital, examines four patients with rare LPNs – Rosai–Dorfman disease, Kimura disease, and Castleman disease – initially misdiagnosed as nonneoplastic conditions. The study aims to underscore the importance of histopathological evaluation in achieving accurate diagnoses and initiating timely management. Results reveal diagnostic complexities, with an average of 2.5 biopsies required for accurate diagnosis.
Conclusions: In-depth case discussions highlight the unique challenges and treatment strategies for each LPN. The central theme emphasizes the critical role of histopathology in distinguishing these rare LPNs from more common musculoskeletal disorders, guiding appropriate therapeutic interventions, and optimizing patient outcomes. These cases serve as poignant examples of the diagnostic pitfalls encountered by clinicians when faced with rare LPNs, reinforcing the necessity of a nuanced approach and collaboration between clinicians and pathologists in achieving accurate diagnoses.
Keywords: Lymphoproliferative neoplasms, musculoskeletal manifestations, histopathological evaluation, diagnostic challenges, rare diseases
Hematologic diseases can lead to a diverse array of musculoskeletal manifestations, including pain, masses, and even fractures. Among those disease, are inflammatory conditions known as lymphoproliferative neoplasms (LPNs) [1]. Although rare, these entities have the potential to induce musculoskeletal pathology. When LPNs affect the musculoskeletal system, they are often mistaken for more common diseases, and difficult to diagnose. History, physical examination, and advanced imaging can aid in diagnosis, but the interpretation of biopsies by experienced pathologists remains pivotal. Rare LPNs such as Rosai-Dorfman disease (RDD), Kimura disease, and Castleman’s disease infrequently manifest with orthopedic chief complaints. Therefore, appropriate and timely diagnosis of such LPNs can prove challenging due to the nonspecific clinical presentation and imaging features. This often results in delayed diagnosis as well as treatment. The aim of the present study was to emphasize the diagnostic challenges associated with LPNs. By retrospectively reviewing our institutions’ experience, we highlight the importance of considering LPNs in the differential diagnosis of atypical musculoskeletal complaints.
This study was performed within the department of orthopedics at a single academic institution. It comprised four patients with rare LPNs, treated at a tertiary care hospital between March 2012 and March 2023. Data and imaging were abstracted manually from the electronic medical record.
The mean age and body mass index at the time of diagnosis were 30 years (range 33) and 24.47 kg/m², respectively. Three patients were male. On an average, 2.5 biopsies were required to obtain an accurate diagnosis. One patient underwent five biopsies, multiple surgical interventions, and radiation treatment prior to the correct diagnosis. Alternative diagnoses included sarcoma, lymphoma, caseating and noncaseating granuloma, osteomyelitis, arteriovenous malformation (AVM), and nodular fasciitis. Three of the four patients underwent surgical intervention by one of the senior authors for definitive diagnosis and treatment (Table 1).
Case 1: Rosai–Dorfman disease
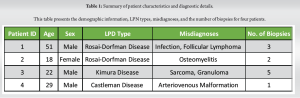
A 51-year-old male with a medical history notable for syndrome of inappropriate antidiuretic hormone and epilepsy presented with left thigh pain. He had initially presented to an outside emergency department 7 months prior, complaining of left lateral and anterior thigh pain. A needle biopsy at that time suggested infection. Three weeks later, femoral reaming without nail placement was performed to debride the suspected infection. Specimens were collected intraoperatively and sent to pathology, at which time the patient was diagnosed with follicular lymphoma and subsequently received 24Gy of external beam radiotherapy. The patient presented to our institution for a second opinion and pathology obtained at the outside institution was deemed to be more consistent with acute on chronic osteomyelitis rather than lymphoma. Two months later, an open biopsy of the femoral lesion was conducted. This led to the development of an abscess at the surgical site, with subsequent identification of a bacterial strain. Treatment for the abscess included a 6-week course of intravenous (IV) antibiotics. The patient’s pain improved with antibiotics and worsened with radiotherapy. In addition, at the time of initial diagnosis over a year and a half prior, there was no evidence of lymphoma elsewhere in the patient’s body. Unfortunately, his pain never resolved completely, and there continued to be notable positron emission tomography (PET) avidity within the majority of the left femoral diaphysis. For this reason, the decision was made to perform repeat open biopsy through a larger cortical window with curettage and placement of antibiotic cement in the intramedullary canal for suspected chronic osteomyelitis. Intraoperative pathology demonstrated histiocytes consistent with RDD. The patient was referred to hematology for further workup and management. The computed tomography (CT)/PET was markedly diminished relative to precurettage. Hematology recommends repeat PET/CT to assess for recurrence and response to radiation. If there is a recurrence, a mitogen-activated protein kinase kinase (MEK) inhibitor such as cobimetinib may be implemented which target cell growth (Fig. 1).
Case 2: Rosai-dorfman disease
An 18-year-old female with a history of cerebral palsy presented with 10 months of left leg pain at rest, which was particularly severe at night. The pain was only partially responsive to nonsteroidal anti-inflammatory drugs and narcotics. Initial evaluation at an outside hospital was felt to be consistent with osteomyelitis and led to her being treated with IV antibiotics for 5 weeks. Outside imaging demonstrated a diaphyseal lytic lesion, with geographic 1B margins. A defect in the cortex at the biopsy site was also noted. There was significant peri-lesional edema extending from the physeal scar to the distal third of the diaphysis, with minimal soft-tissue edema surrounding the biopsy track. Due to the imaging appearance, a biopsy was performed and found consistent with RDD. Initial treatment at an outside institution involving unknown IV steroids temporarily alleviated the patient’s symptoms, but the pain recurred. She required repeated administration of IV steroids and morphine. The patient had no history of similar episodes. Her symptoms were refractory to conservative management, and as such, extended curetting and tibial bone grafting were recommended. She tolerated the procedure without event. A whole-body CT was done 4 months later, showing some increased fludeoxyglucose activity in the proximal left tibia, suggesting some residual disease. Radiographs of the left tibia taken at the same time showed incorporation of the bone graft at the location of the cortical window, with no further lucencies. She was then seen by hematology, who began treatment with 6-MP 100 mg daily, and methotrexate 30 mg weekly. She subsequently underwent a repeat curettage of the lesion, and has had subsequent healing with reduction in pain.
Case 3: Kimura disease
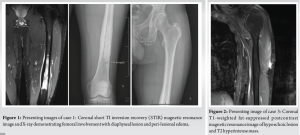
A 22-year-old male presented with a painless mass at the insertion of his left deltoid, progressively growing over 2 months. No associated symptoms were reported. There was no history of trauma or local injections at the site. Physical examination revealed a painless mass with no other lesions noted. Ultrasound performed at the initial visit demonstrated a 1.1 cm × 0.5 cm × 1 cm serpiginous-shaped hypoechoic lesion with no overt internal vascularity. Magnetic resonance imaging (MRI) further characterized the mass, demonstrating a lobulated T2 hyperintense avidly enhancing mass within the subcutaneous fat of the lateral, left upper extremity that had infiltrating tails involving the deltoid muscle bellies. The initial differential diagnosis included sarcoma and granuloma in the setting of prior injection or nodular fasciitis. An ultrasound-guided biopsy was recommended for definitive diagnosis. Histologic sections of core biopsy demonstrated needle-shaped fibroadipose tissue with lymphocytic proliferation, favoring reactive lymphoid proliferation without sufficient morphologic evidence of lymphoma. Additional B- and T-cell clonality polymerase chain reaction testing was recommended for further assessment. Immunohistochemistry was performed and the presence of increased mast cells expressing CD117 and increased Langerhans’ cells expressing CD1a was confirmed. Although increased, mast cells and Langerhans’ cells are mostly scattered and do not form abnormal sheets or clusters, arguing against the presence of mastocytosis and of Langerhans cell histiocytosis, respectively. CD25 and CD2 highlight small lymphoid cells and appear negative in mast cells. Tryptase highlights a subset of mast cells. In situ hybridization for Epstein-Barr virus-encoded RNA (EBER) is negative. The patient subsequently underwent excision of the mass in his left upper extremity. The intraoperative pathology report of fibroadipose tissue with predominant eosinophilia favored the diagnosis of secondary Kimura disease. The patient tolerated the aforementioned procedure well. Subsequent follow-up in the rheumatology clinic showed the patient to be symptom-free after surgical excision with no recurrence on the site, lymphadenopathy, or new lesions. They follow-up with annual laboratories and monitor clinically (Fig. 2).
Case 4: Castleman disease
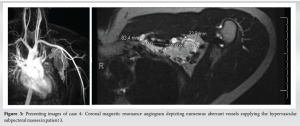
An otherwise healthy 29-year-old male with no comorbidities and no surgical history was referred to our clinic due to the incidental discovery of a left axillary mass on self-examination, initially concerning AVM. No prior symptoms or discomfort associated with the mass were reported. Ultrasound demonstrated a clustered vascular lesion with mostly venous flow measuring 9.4 cm ×3.16 cm ×5.33 cm, consistent with an arterio-venous malformation. MRI revealed multiple hypervascular left subpectoral/supraclavicular masses, the largest and most dominant measuring up to 8.5 cm in the subpectoral region. Multiple peritumoral and intratumoral feeding arteries and draining veins were identified. The differential diagnosis included AVM, alveolar soft part sarcoma, angiosarcoma, or hypervascular metastases. A biopsy was recommended to exclude more malignant etiologies. Subsequent fine-needle aspiration of the mass revealed a predominance of blood with scattered lymphoid aggregates indicative of lymphangioma. Following an open, excisional biopsy, the final diagnosis was determined to be Castleman disease. The clinical course was characterized by successful excision of the left axillary mass, coupled with ongoing surveillance and a proactive approach to long-term management with hematology/oncology. During a follow-up appointment 6 months after the initial presentation, the patient reported significant improvement postexcisional biopsy, with a return to his usual activities. He experienced some residual symptoms, such as fatigue and numbness in the axillary region. All the laboratories were normal, imaging demonstrated no additional lesion. Hematology did not have to proceed with therapy (Fig. 3).
LPNs constitute a complex and diverse group of disorders characterized by the uncontrolled proliferation of lymphocytes or their precursors. These conditions can manifest with a wide spectrum of clinical presentations, frequently resembling other nonneoplastic disorders. In this case series, we present four distinct cases of patients initially misdiagnosed with nonneoplastic conditions but ultimately found to have rare LPNs – Kimura disease, Castleman disease, and RDD. This discussion explores the diagnostic challenges posed by these conditions and underscores the importance of histopathological evaluation in achieving accurate diagnoses. The pathogenesis of LPNs involves underlying lymphocyte development and homeostasis dysfunction [2]. Typically, LPNs manifest as lymphocyte accumulation in the bone marrow, peripheral blood, or lymph nodes, and spleen, rather than presenting as orthopedic complaints2. Our series of rare LPN cases underscores their unique and atypical manifestations and treatment strategies, as discussed here.
Rosai–Dorfman disease
RDD is a rare nonlangerhans cell histiocytosis with variable clinical presentations. The pathophysiology remains poorly defined but is believed to be associated with neoplastic, infectious, or autoimmune processes [3]. Extranodal involvement occurs in 43% of cases, with bone involvement observed in <10% [3,4]. Typically, this disease affects younger patients, with a median age of diagnosis around 20 years, and presents as painless cervical adenopathy with constitutional symptoms [3]. The imaging manifestations of bone involvement are nonspecific, necessitating histopathological examination with immunohistochemistry for confirmation [4]. Key features include pale cytoplasm and histiocytes demonstrating emperipolesis [3]. Immunophenotypic markers supporting the diagnosis include S-100, fascin, and CD68 positivity in affected histiocytes, distinguishing RDD from Langerhans histiocytosis [3]. RDD is often initially misdiagnosed as chronic osteomyelitis or giant cell tumor based on imaging findings, but histological and immunohistochemical analyses facilitate accurate diagnosis [5]. While treatment approaches vary, surgical surgical resection or curettage are frequently employed [3]. In our case, the patient’s initial misdiagnosis as follicular lymphoma based on imaging findings led to radiation therapy. Subsequent evaluation and histopathological analysis revealed RDD, emphasizing the diagnostic challenges and potential consequences of mistaking RDD for other neoplastic entities. Surgical resection or curettage is commonly employed for RDD treatment, further highlighting the importance of correct diagnosis.
Kimura disease
Kimura disease is a rare, chronic inflammatory disorder of unknown etiology [6]. Kimura’s disease is considered a Type I allergy due to elevated eosinophil counts and immunoglobulin E (IgE) levels in blood, but its precise pathogenesis remains unknown [6,7]. It often manifests as a painless soft tissue mass, primarily in the head and neck, followed by the axilla, groin, popliteal region, and arm [7]. It predominantly affects men in their second or third decade of life and is characterized by elevated eosinophil counts and IgE levels [6]. Diagnosis relies on histological analysis demonstrating eosinophil infiltration with follicular hyperplasia, fibrocollagenous proliferation, and vascular proliferation of postcapillary venules [6]. Treatment modalities include surgical excision, steroid therapy, and radiotherapy, though no standardized approach exists [7]. In our case, the initial inconclusive imaging findings raised concerns about sarcoma or granuloma. Still, a targeted ultrasound-guided biopsy ultimately unveiled Kimura’s disease, leading to an appropriate course of action. This underscores the importance of considering rare entities like Kimura disease in the differential diagnosis, particularly when presented with unusual soft tissue masses in atypical locations.
Castleman disease
Castleman disease is a rare, chronic lymphoproliferative disorder of unknown etiology, with an incidence of 1/50,000 [8]. It typically presents as a mediastinal nodal mass or, in the extranodal form, as masses in the mediastinum or retroperitoneum [9]. It is characterized by lymphoid hyperplasia and has two variants: the hyaline vascular type and the plasma cell type [10]. Castleman disease rarely presents in the extremities [9,10]. Diagnosis of Castleman disease can be based on histopathological features, including peripheral fat, medium-sized vessels at the lesion’s periphery, and central hypovascularity [9]. Characteristic imaging features include peripheral fat, central calcifications, and prominent peripheral vasculature [9]. Despite its rarity, providers should be vigilante for Castleman disease due to its potential for transformation into follicular dendritic cell sarcoma [9]. In our case, the initial ultrasound suggested AVM, yet a subsequent excisional biopsy unmasked Castleman disease. The importance of this case lies in the need for a thorough evaluation when encountering hypervascular masses and considering Castleman disease even in extranodal sites. Early recognition can prevent inappropriate treatment and guide proper management. The central theme in all three cases presented here is the pivotal role of histopathological evaluation by experienced pathologists. Misdiagnosing LPNs can have significant consequences, including delayed treatment, inappropriate management, and potentially adverse outcomes. The cases presented in this series serve as poignant examples of the difficulties clinicians face when encountering rare LPNs, as these conditions often mimic more common and benign musculoskeletal disorders. Histopathology allows for the identification of characteristic features that differentiate LPNs from their mimics. In Kimura disease, it confirms eosinophil infiltration, follicular hyperplasia, and vascular proliferation. In Castleman disease, it reveals lymphoid hyperplasia with specific vascular patterns. For RDD, it demonstrates histiocytes with emperipolesis and specific immunophenotypic markers. In the realm of rare LPNs, where clinical and imaging features often overlap with more common conditions, the ability to discern subtle histological distinctions becomes paramount.
The cases in this series serve as reminders of the diagnostic challenges posed by rare LPNs such as Kimura disease, Castleman disease, and RDD. Their ability to mimic more common conditions necessitates a high index of suspicion among clinicians, coupled with the expertise of pathologists to accurately differentiate these neoplasms. Timely and precise diagnosis is essential for initiating appropriate management, thereby optimizing patient outcomes and avoiding the pitfalls of misclassification.
Rare LPNs such as RDD, Kimura disease, and Castleman disease often present with atypical musculoskeletal symptoms, leading to diagnostic challenges and potential misdiagnoses. These rare LPNs often mimic common conditions, causing significant diagnostic difficulties. Accurate diagnosis of LPNs is essential for appropriate treatment, as correct identification avoids inappropriate management and optimizes patient outcomes. The key takeaway is the critical importance of thorough histopathological evaluation in distinguishing these rare neoplasms from more common conditions. By highlighting these diagnostic challenges, the report underscores the need for heightened awareness and a methodical diagnostic process to ensure timely and appropriate treatment for optimal patient outcomes.
References
- 1.Morais SA, du Preez HE, Akhtar MR, Cross S, Isenberg DA. Musculoskeletal complications of haematological disease. Rheumatology (Oxford) 2016;55:968-81. [Google Scholar | PubMed]
- 2.Sands J, Tuscano JM. Geoepidemiology and autoimmune manifestations of lymphoproliferative disorders. Autoimmun Rev 2010;9:A335-41. [Google Scholar | PubMed]
- 3.Moore CE, Flint JH, Taniguchi KM, Gable PS. Rosai-dorfman: Rare manifestations of a rare disease. Cureus 2023;15:e36673. [Google Scholar | PubMed]
- 4.Sassi F, M'rad H, Kacem LB, Sassi B, Hannachi S, Rammeh S. A lesion of the patella: An unexpected location of Rosai-dorfman disease: A case report. Int J Surg Case Rep 2022;98:107510. [Google Scholar | PubMed]
- 5.Baker JC, Kyriakos M, McDonald DJ, Rubin DA. Primary Rosai-dorfman disease of the femur. Skeletal Radiol 2017;46:129-35. [Google Scholar | PubMed]
- 6.Chung YG, Jee WH, Kang YK, Jung CK, Park GS, Lee AH, et al. Kimura’s disease involving a long bone. Skeletal Radiol 2010;39:495-500. [Google Scholar | PubMed]
- 7.Morinaga S, Yamamoto N, Hayashi K, Takeuchi A, Miwa S, Igarashi K, et al. Kimura’s disease diagnosed in the department of orthopedic surgery treated with wide excision: Report of two cases. In Vivo 2023;37:1373-8. [Google Scholar | PubMed]
- 8.Liu SZ, Zhou X, Wang YP, Liu Y. Osteosclerosis in Castleman’s disease. QJM 2022;115:171-2. [Google Scholar | PubMed]
- 9.Eward WC, DeWitt SB, Brigman BE, Kontogeorgakos V, Lagoo AS. Extranodal Castleman disease of the extremities: A case report and review of the literature. Skeletal Radiol 2014;43:1627-31. [Google Scholar | PubMed]
- 10.Rooney RC, Pitcher JD. Castleman’s disease in the extremity. Am J Orthop (Belle Mead NJ) 1998;27:373-4. [Google Scholar | PubMed]






Related Articles in Journal of Orthopaedic Case Reports
 August 1, 2025 Acute Carpal Tunnel Syndrome: Is It Only Wrist Fractures? A Case Series Report and Literature Review
August 1, 2025 Acute Carpal Tunnel Syndrome: Is It Only Wrist Fractures? A Case Series Report and Literature Review November 10, 2018 Clavicular Osteochondroma: Extremely Rare Cause of Impingement Syndrome
November 10, 2018 Clavicular Osteochondroma: Extremely Rare Cause of Impingement Syndrome May 10, 2021 Wedge Osteotomy with Tens Nailing in Monostotic Fibrous Dysplasia of Tibia – A Case Report
May 10, 2021 Wedge Osteotomy with Tens Nailing in Monostotic Fibrous Dysplasia of Tibia – A Case Report July 7, 2017 Tension Band Wiring Is As Effective As A Compression Screw In A Neglected, Medial Maleolus Non-Union: A Case-Based Discussion & Literature Review
July 7, 2017 Tension Band Wiring Is As Effective As A Compression Screw In A Neglected, Medial Maleolus Non-Union: A Case-Based Discussion & Literature Review