
A rapidly growing cranial swelling in children aged 5–15 years should raise suspicion for Ewing sarcoma of the skull. Definitive diagnosis relies on imaging studies followed by histopathological analysis. The treatment of Ewing sarcoma in this location is complex and requires a multidisciplinary approach, incorporating chemo-radiotherapy and surgical resection.
Dr. Rishabh Gupta, Resident, Deptt of Orthopedics Dr Rajendra Prasad Government Medical College, Tanda, Himachal Pradesh, India. E-mail: docvipinsharma@gmail.com
Introduction: Ewing sarcoma of the skull (EWS) is an exceptionally rare variant, accounting for approximately 1% of all Ewing sarcoma cases. Due to its rarity, there are only a limited number of documented instances in medical literature, making it a topic of significant interest and importance in the field of oncology.
Case Report: A 13-year-old boy presented with a gradually enlarging swelling measuring 5 × 4 cm in the left parietotemporal region. Histopathological analysis confirmed a diagnosis of Ewing sarcoma of the cranium. The patient underwent neoadjuvant chemotherapy and radiotherapy, followed by surgical resection. However, 2 years later, he experienced a recurrence characterized by exophytic growth and intracranial involvement. Although a revised course of chemoradiotherapy was planned, the patient succumbed to his condition on the 14th day of hospitalization.
Conclusion: The definitive diagnosis in such cases is often complex and relies heavily on histopathological findings. Early detection, along with prompt multidisciplinary intervention, is essential for effective management and improved patient outcomes.
Keywords: Chemotherapy, ewing sarcoma, rare, skull.
Ewing sarcoma is a malignant tumor commonly affecting children and adolescents aged 5–15 years, typically involving the metadiaphyseal regions of long bones, flat bones, and soft tissues. Ewing sarcoma of the skull (EWS) is rare, with an incidence of 1–6% [1]. We present a case of primary EWS in a 13-year-old male, who initially presented with swelling in the left parietotemporal region. The patient underwent neoadjuvant chemotherapy and radiotherapy followed by surgical resection. Two years later, he experienced a recurrence characterized by exophytic growth and intracranial involvement, which ultimately led to his demise.
A 13-year-old boy presented with a hard, non-tender swelling in the left parietotemporal area, initially measuring 5 × 4 cm, which gradually increased to 10 × 7 cm (Fig. 1). X-ray imaging suggested lytic permeative destruction with a sunburst appearance of the outer table (Fig. 2) with magnetic resonance imaging (MRI) showing exophytic growth of the tumor. Histopathological examination of the lesion revealed a blue round cell tumor that was positive for CD99 (Fig. 3). The patient subsequently underwent neoadjuvant chemotherapy and radiotherapy, followed by surgical resection. He remained well for 2 years but was readmitted with status epilepticus and a high-grade fever. Upon admission, he exhibited fever and seizures, with the fever likely resulting from hypothalamic dysfunction due to the local spread of Ewing’s sarcoma. Seizures were managed with Levetiracetam, Sodium Valproate, and Phenytoin. Clinically, there was a recurrence of growth in the left parietotemporal area, now measuring 15 × 10 × 16 cm, larger than the original tumor. A complete blood count indicated anemia, while other blood tests, including serum uric acid, phosphorus, calcium, and electrolytes, were within normal limits. MRI of the brain revealed an exophytic growth of 15 × 10 × 16 cm, with intracranial extension involving the left parietotemporal region, a midline shift of 2.2 cm, and left uncal herniation accompanied by extensive perilesional edema in the left cerebral white matter (Fig. 4a and b). Despite planning a revised course of radiotherapy and chemotherapy, the patient succumbed on the 14th day of admission.
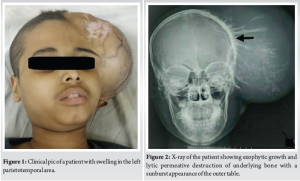
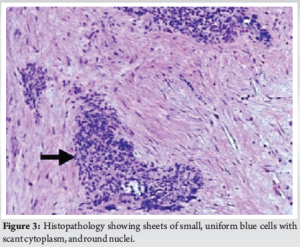
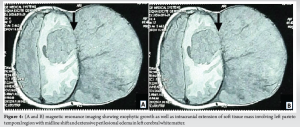
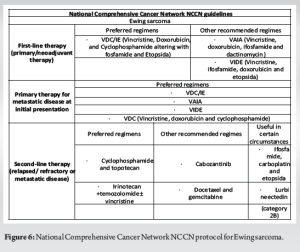
The current study examines a unique case of EWS of the cranium, initially manifesting as an extracranial growth in the left parietotemporal region. This case later progressed to local intracranial spread, affecting the left cerebral hemisphere and resulting in leucomalacia, mass effect, and midline shift, ultimately leading to uncal herniation. To date, approximately 80 cases of EWS involving the cranium have been documented in the literature, with around 80% of these tumors occurring in the supratentorial region, making posterior fossa presentations relatively uncommon [2]. The most frequently affected areas of the skull vault include the frontal (28%), parietal (22%), and temporal bones (11%), while the involvement of the ethmoid, petrous, orbital, and maxillary bones is rare [3]. Metastasis from primary EWS of the skull is infrequent compared to skeletal presentations [4]. The origin of EWS remains elusive. James Ewing proposed an endothelial origin in 1921, but subsequent findings suggesting myelocyte-like ultrastructural features shifted focus toward a myelogenous theory [5]. Evidence of neuroectodermal origin is supported by the expression of markers such as neuron-specific enolase and S-100, while CD99-a membrane protein, serves as a relatively specific marker for EWS, and is detected at low levels in mesenchymal stem cells [6]. EWS primarily affects children and adolescents aged 5–15, with only 6.3% of cases identified in individuals over 50. Older age is associated with poorer prognostic outcomes due to larger, more advanced, and invasive tumors at diagnosis [2]. EWS of the skull is quite rare, accounting for just 1–2% of cases, and can originate from various locations, including the petrous, occipital, parietal, temporal, and sphenoid bones, often exhibiting poor margination and significant soft tissue extension (80%) [7]. In the present case, the EWS was seen to arise from the left parietotemporal bone, exophyting to the exterior, and spreading intracranially into left cerebral white matter, with a midline shift to the right with left uncal herniation. Radiologically, EWS is characterized by permeative destruction, mottling, erosions, and a lamellated periosteal reaction [8], although less common features such as the Codman triangle and sunburst appearance may also be observed. Sometimes EWS may present as osteolytic lesions in the skull and it is crucial to differentiate it from identical-looking lesions, such as eosinophilic granuloma, metastases, Hand Schuller Christian syndrome, Burkitt’s lymphoma, Fibrous Dysplasia, Aneurysmal bone cyst, Osteoclastoma, and Giant cell reparative granuloma. Patterns of destruction, presence of soft tissue, calcification, cystic component, and septations help differentiate among these lesions radiologically [9]. In the present case, imaging revealed lytic permeative destruction and a sunburst appearance from the outer table of the left temporoparietal bone. MRI indicated a 15 × 10 × 16 cm exophytic tumor with intracranial involvement and associated mass effect. Although local radiology and MRI have a role to play, a definitive diagnosis was achieved through core needle biopsy, which demonstrated round-to-oval cells arranged in lobules separated by a thin vascular channel, vesicular nuclei and indistinct nucleoli with characteristic mitotic figures, and focal necrosis. Immunohistochemical staining confirmed positivity for CD99 and solidifying the diagnosis of EWS. Other than EWS, blue round cell tumors can be found in lymphoma, malignant melanoma, rhabdomyosarcoma, olfactory neuroblastoma, undifferentiated carcinoma, histopathology, and subsequent immunohistochemistry is helpful in confirmation of diagnosis [10]. In the present case, CD 99 positivity confirmed the diagnosis. Staging is essential for assessing the extent of disease spread. Traditional imaging techniques such as computed tomography (CT), MRI, and bone scans are limited in their ability to differentiate between viable, necrotic, and residual tumors. However, 18F-fluorodeoxyglucose positron emission tomography-CT combines anatomical and metabolic imaging defines the glycolytic metabolism of tumor cells [11] (Fig. 5). In the present case, moderate-to-high uptake in the left parietotemporal region and left cerebral hemisphere in this case. EWS of the skull can invade the brain causing mass effect, local spread from the inner table of the skull, or distant metastasis. Mass effect results in obstructive hydrocephalus leading to headaches, nausea, and vomiting. Although brain metastases are rare in pediatric solid tumors, EWS and Rhabdomyosarcoma are among those most likely to spread to the brain [12]. In this instance, the tumor exhibited local brain invasion, resulting in a left-to-right midline shift. Survival rates for EWS have improved with multimodal treatment, including surgery, radiotherapy, and chemotherapy. Non-metastatic cases report 5-year survival rates of up to 60%, whereas metastatic presentations yield only 10%. In non-metastatic cases of Ewing Sarcoma, neoadjuvant chemotherapy consisting of vincristine, cyclophosphamide, doxorubicin, alternating with ifosfamide and etoposide is administered for 12–15 cycles over a period of 8–12 months. This treatment is followed by surgical resection with adequate oncological margins. Subsequent adjuvant radiotherapy and maintenance chemotherapy are recommended. For patients diagnosed with metastatic disease, the same standardized chemotherapy regimen is utilized as for localized cases (Fig. 6). In instances where patients with lung metastases achieve complete remission, total lung irradiation may be considered. In addition, supplemental irradiation is typically indicated for bone metastases. For those experiencing systemic or local relapse, palliative treatment options are explored [13]. In this case, the patient received neoadjuvant chemotherapy for 8 weeks, followed by tumor excision and adjuvant chemotherapy [14] with the regimen of Cyclophosphamide, Vincristine, Doxorubicin alternating with ifosfamide and etoposide. Unfortunately, the disease recurred 2 years later with both extracranial and intracranial involvement, leading to palliative care considerations, but the patient succumbed shortly thereafter.
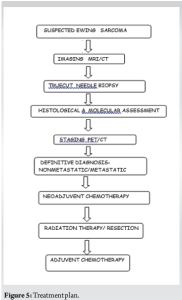
Ewing’s sarcoma presenting as a primary cranial lesion is a rare phenomenon. Diagnosing such cases can be particularly challenging and necessitates comprehensive immunohistological analysis to exclude various differential diagnoses. Early and accurate diagnosis, followed by prompt multidisciplinary intervention, is essential for improving outcomes. The preferred treatment approach involves primary surgical excision with clear margins, followed by multiagent adjuvant chemotherapy and local radiotherapy. When initiated early in the disease course, this optimal treatment regimen can significantly enhance prognosis.
In children aged 5–15 years, the presence of a rapidly enlarging cranial mass should prompt consideration of EWS. Accurate diagnosis necessitates a combination of imaging studies and histopathological evaluation. Managing Ewing sarcoma in this anatomical site is intricate and demands a multidisciplinary strategy that includes chemotherapy, radiotherapy, and surgical intervention.
References
- 1.Sharma AD, Singh J, Bhattacharya J. Primary Ewing’s sarcoma of cranium in a pediatric patient. J Pediatr Neurosci 2017;12:273-5. [Google Scholar | PubMed]
- 2.Ravina K, Windermere SA, Zhao Q, Lerner A, Dyer M, Upadhyay U, et al. Primary intracranial extraosseous Ewing’s sarcoma of the skull base in an elderly adult: Illustrative case. J Neurosurg Case Lessons 2022;4:CASE22214. [Google Scholar | PubMed]
- 3.Kadar AA, Hearst MJ, Collins MH, Mangano FT, Samy RN. Ewing’s sarcoma of the petrous temporal bone: Case report and literature review. Skull Base. 2010;20:213-7. [Google Scholar | PubMed]
- 4.Falk S, Alpert M. Five-year survival of patients with Ewing’s sarcoma. Surg Gynecol Obstet 1967;124:319-24. [Google Scholar | PubMed]
- 5.Kadin ME, Bensch KG. On the origin of Ewing’s tumor. Cancer 1971;27:257-73. [Google Scholar | PubMed]
- 6.Tu J, Huo Z, Gingold J, Zhao R, Shen J, Lee DF. The histogenesis of ewing sarcoma. Cancer Rep Rev. 2017;1:10.15761/CRR.1000111. [Google Scholar | PubMed | CrossRef]
- 7.Wang D, Guo Z. Multiple primary Ewing’s sarcomas in cerebral cranium of a child: A case report and review of the literature. Int J Clin Exp Pathol 2015;8:7575-82. [Google Scholar | PubMed]
- 8.Jin TY, Gaillard F. Ewing Sarcoma. Radiology Case. Available from: https://www/radiopaedia.org/articles/ewing-sarcoma Last accessed on 3 Nov 2024 [Google Scholar | PubMed]
- 9.Gadani S, Mody RP, Solanki RN, Mahajan A. Primary Ewing sarcoma of skull vault in a child. Indian J Radiol Imaging 2003;13:303-5. [Google Scholar | PubMed]
- 10.Thorn D, Mamot C, Krasniqi F, Metternich F, Prestin S. Multimodality treatment in Ewing’s sarcoma family tumors of the maxilla and maxillary sinus: Review of the literature. Sarcoma 2016;2016:3872768. [Google Scholar | PubMed]
- 11.Guimarães JB, Rigo L, Lewin F, Emerick A. The importance of PET/CT in the evaluation of patients with Ewing tumors. Radiol Bras 2015;48:175-80. [Google Scholar | PubMed]
- 12.Parasuraman S, Langston J, Rao BN, Poquette CA, Jenkins JJ, Merchant T, et al. Brain metastases in pediatric Ewing Sarcoma and rhabdomyosarcoma: The St. Jude Children’s Research Hospital experience. J Pediatr Hematol Oncol 1999;21:370-7. [Google Scholar | PubMed]
- 13.Paulussen M, Bielack S, Jürgens H, Casali PG, ESMO Guidelines Working Group. Ewing’s sarcoma of the bone: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 2009;20 Suppl 4:140-2. [Google Scholar | PubMed]
- 14.Bhattacharjee S, Venkata SR, Uppin MS. Skull and spinal Ewing’s sarcoma in children: An institutional study. J Pediatr Neurosci 2018;13:392-7. [Google Scholar | PubMed]



Related Articles in Journal of Orthopaedic Case Reports
 August 1, 2025 Melorheostosis: A Rare Case Report with Pelvic and Lumbar Involvement
August 1, 2025 Melorheostosis: A Rare Case Report with Pelvic and Lumbar Involvement June 1, 2025 Successful Management of Atypical Bilateral Galeazzi Fractures with Unique Dorsal Displacement: A Case Report
June 1, 2025 Successful Management of Atypical Bilateral Galeazzi Fractures with Unique Dorsal Displacement: A Case Report April 1, 2025 Tenosynovial Giant Cell Tumor Involving Quadriceps Muscle – A Case Report of Two Cases
April 1, 2025 Tenosynovial Giant Cell Tumor Involving Quadriceps Muscle – A Case Report of Two Cases January 1, 2025 Atraumatic Bilateral Transcervical Femoral Neck Fractures in an Elderly Epileptic Patient: A Case Report
January 1, 2025 Atraumatic Bilateral Transcervical Femoral Neck Fractures in an Elderly Epileptic Patient: A Case Report