
The importance of considering Phosphaturic Mesenchymal Tumors (PMTs) in patients presenting with unusual clinical symptoms and the effectiveness of surgical management in achieving favorable outcomes.
Dr. Sharafuddeen Mammu, Department of orthopaedic surgery, Government Medical College, Kozhikode, Kerala, India. E-mail: Sharafuddeen786@gmail.com
Abstract
Introduction: Phosphaturic mesenchymal tumors (PMTs) are rare bone neoplasms with diverse clinical presentations, often posing diagnostic challenges.
Case Report: We describe the case of a 37-year-old female schoolteacher with a PMT localized in the distal femur. Diagnostic indicators included hypophosphatemia, hyperphosphaturia, elevated fibroblast growth factor-23 levels, and clinical symptoms of osteomalacia. Surgical management involved tumor resection and limb salvage surgery with a megaprosthesis. The post-operative period was uneventful, leading to a stable discharge. On follow-up, the patient showed no signs of recurrence, regained full ambulation, remained pain-free, and resumed teaching comfortably.
Conclusion: This case highlights the importance of considering PMT in patients with unusual clinical symptoms, accompanied by hypophosphatemia, hyperphosphaturia, and osteomalacia, and demonstrates successful surgical management, leading to a favorable outcome.
Keywords: Phosphaturic mesenchymal tumor, osteomalacia, hypophosphatemia, hyperphosphaturia, fibroblast growth factor 23.
Phosphaturic mesenchymal tumors (PMTs) usually occur in bones and other connective tissue. The characteristic feature is renal phosphate wasting which leads to the phenomenon of tumor-induced osteomalacia marked by a reduction in osteoblastic activity [1]. Tumor-induced osteomalacia (TIO) is caused due to the secretion of fibroblast growth factor (FGF) 23, which is a peptide which acts on kidneys resulting in excessive excretion of phosphate and reduced renal reabsorption. These are slow growing and benign in nature although it can cause significant physical discomfort and impairment. Despite advances in diagnostic imaging modalities and histopathological techniques, PMT remains a diagnostic conundrum due to its nonspecific clinical presentation and radiographic features, often mimicking more common bone tumors or metabolic bone diseases. The diagnosis and locating the responsible tumors can be challenging and tumor is often missed or misdiagnosed [2]. The challenge in accurately diagnosing PMT lies in its rarity and the variable presentation of TIO, which can include bone pain, fractures, muscle weakness, and even pathological fractures, thus leading to a diagnostic odyssey for patients and clinicians alike. Here, we present a case of PMT of the distal femur which posed significant diagnostic challenges.
A 37-year-old female schoolteacher has been experiencing left thigh pain for the past 2 years, significantly affecting her profession and activities of daily living for the past 6 months. Clinical examination reveals diffuse fullness on the anterolateral aspect of the left thigh, along with atrophy in the thigh and calf muscles. In addition, there is a fixed flexion deformity of 10° in the left knee, without any signs of instability.
The patient’s laboratory results include a white blood cell (WBC) count of 9200, an erythrocyte sedimentation rate (ESR) of 75 mm in 1 h, serum calcium level of 7.8 mg/dL (reference range: 8.4–11), serum phosphorus level of 2 mg/dL (reference range: 2.5–4.5), and an alkaline phosphatase level of 161. The 1,25 dihydroxycholecalciferol level is 18, and a 24-h urine phosphorus test reveals 1388 mg (reference range: 400–1300 mg/day). Radiological examinations, including X-ray and magnetic resonance imaging (MRI), indicate a lytic lesion in the distal femur with specific dimensions and features. Technitium 99 bone scintigraphy reveals increased osteoblastic activity and positron emission tomography-computed tomography identified a primary malignant lesion expressing somatostatin receptor in the distal end of the femur, suggestive of a mesenchymal tumor (Fig. 1). The tru-cut biopsy results confirm a PMT with marked nuclear atypia and low mitotic activity. Immunohistochemistry shows positive staining for special AT-rich sequence-binding protein 2 (SATB2) and CD 56.
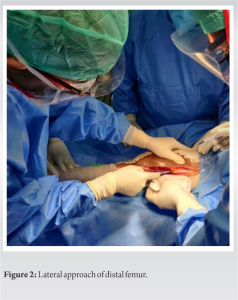
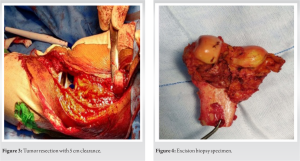
After all radiological workup and tru-cut biopsy, limb salvage surgery was planned. The operative procedure was done through a lateral approach (Fig. 2), resection of the tumor with 5 cm clearance was done (Fig. 3), implanted with Adler (RESTOR) megaprosthesis and specimen sent for biopsy (Fig. 4).
The patient was mobilized next day using walking frame as tolerated. Deep vein thrombosis prophylaxis was given for 2 weeks. Histopathological examination confirmed the diagnosis with bland spindle cells with grungy calcification (Fig. 5).
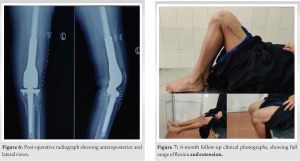
On 6-month follow-up, no further lesions were identified, radiographs showed a well-fitting implant (Fig. 6). The patient was ambulant without support, pain free doing her day-to-day activities (Fig. 7).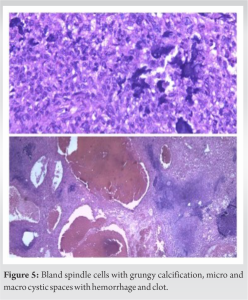
PMTs are uncommon and are often found out clinically when it manifests as a paraneoplastic syndrome, resulting in TIO. PMTs are relatively rare, which posits it with significant diagnostic challenges and should be dealt with a high index of suspicion. The pathogenesis of osteomalacia in PMT is secondary to the production of phosphate-regulating substances by the tumor cells, most importantly the FGF-23. FGF-23 diminishes the proximal renal tubules reabsorption of phosphate and increases renal excretion of the phosphate. One of the most common causes of TIO is found to be PMTs [3]. It was in 1987 that Weidner introduced a classification of tumors causing osteomalacia, based on histological types into connective tissue, osteoblastoma such as, non-ossifying fibroma like subtype and ossifying fibroma like subtype [4]. Most of these are encountered in the long bones of the lower limbs, such as the femur and tibia [5]. It may occur anywhere in the body including soft tissue and bone, thigh, foot, hand, waist, hips, head and neck, and back [6]. In the case presented, the lesion was on the distal femur. The clinical manifestations include fatigue, muscle weakness, bone pain, and spontaneous fractures [2]. In our case, the patient presented with bone pain and inability to walk. Early identification can lead to a significant reduction in morbidity and better outcomes. Locating the tumor poses a significant diagnostic hurdle and may require comprehensive imaging techniques such as total body MRI, computed tomography, and scintigraphy with radiolabeled somatostatin analogs [7]. CT is useful in localizing TIO lesions missed by conventional imaging [8]. Rare cases of metastasis to lung have been reported. Uchihashi et al. described two cases of PMTs resulting in fatal multiple lung metastases [9]. Hence, it is essential to keep a close watch on further emergence of symptoms. In the case presented tumor was located in the distal femur, with clinical features of bone pain, difficulty in walking, investigation showed low calcium, low serum phosphorous, low 1,25 dihydroxy Vitamin D, and increased urinary phosphate excretion. Tru-cut biopsy and histopathology of our case showed PMT with significant nuclear atypia and low mitotic activity. IHC positive for SATB2 and CD 56. Biochemical and urine values were suggestive of FGF-mediated hypophosphatemia. No evidence of metastatic disease was found on the work up. In 6 months of following, no further emergence of symptoms and serum phosphorus levels were normal. The prognosis for benign PMTs is excellent after complete surgical resection. For unresectable or incompletely resected tumors, curative intended radiotherapy is an auxiliary therapeutic option, phosphate, and active Vitamin D supplements are also required [10]. Recent studies have reported that a humanized anti-FGF23 antibody therapy may be useful for TIO patients, and octreotide has been found useful for the regulation of phosphate metabolism [11]. Research on PMT, it is molecular and genetic characterization is still in progress. Due to the debilitating repercussions on bone metabolism, a multidisciplinary team of endocrinologist, oncologist, and orthopedic doctor is crucial for effective management and follow-up.
PMTs represent a rare category of bone disease. Diagnosis is often delayed. Noteworthy diagnostic indicators encompassed hypophosphatemia, hyperphosphaturia, and elevated FGF-23 levels, coupled with clinical manifestations. A multidisciplinary team of endocrinologist, orthopedician, oncosurgeon, and pathologist is essential for the best outcome. A thorough 6-month follow-up in this case revealed an absence of recurrence, underscoring the efficacy of the surgical approach in managing PMT and highlighting the importance of vigilant clinical observation in cases presenting with the biochemical and symptomatic profile of hypophosphatemia.
This case signifies the need for a high index of suspicion for the possibility of rare diagnoses like PMT and the need for a multidisciplinary approach for early diagnosis, and timely intervention can improve patient’s quality of life significantly.
References
- 1.Ghorbani-Aghbolaghi A, Darrow MA, Wang T. Phosphaturic mesenchymal tumor (PMT): Exceptionally rare disease, yet crucial not to miss. Autops Case Rep 2017;7:32-7. [Google Scholar | PubMed]
- 2.Zuo QY, Wang H, Li W, Niu XH, Huang YH, Chen J, et al. Treatment and outcomes of tumor-induced osteomalacia associated with phosphaturic mesenchymal tumors: Retrospective review of 12 patients. BMC Musculoskelet Disord 2017;18:403. [Google Scholar | PubMed]
- 3.Weidner N, Santa Cruz D. Phosphaturic mesenchymal tumors. A polymorphous group causing osteomalacia or rickets. Cancer 1987;59:1442-54. [Google Scholar | PubMed]
- 4.Seijas R, Ares O, Sierra J, Pérez-Dominguez M. Oncogenic osteomalacia: Two case reports with surprisingly different outcomes. Arch Orthop Trauma Surg 2009;129:533-9. [Google Scholar | PubMed]
- 5.Dadoniene J, Miglinas M, Miltiniene D, Vajauskas D, Seinin D, Butenas P, et al. Tumour-induced osteomalacia: A literature review and a case report. World J Surg Oncol 2016;14:4. [Google Scholar | PubMed]
- 6.Qiu S, Cao LL, Qiu Y, Yan P, Li ZX, Du J, et al. Malignant phosphaturic mesenchymal tumor with pulmonary metastasis: A case report. Medicine (Baltimore) 2017;96:e6750. [Google Scholar | PubMed]
- 7.Beygi S, Denio A, Sharma TS. The foot that broke both hips: A case report and literature review of tumor-induced osteomalacia. Case Rep Rheumatol 2017;2017:3191673. [Google Scholar | PubMed]
- 8.Dutta D, Pandey RK, Gogoi R, Solanki N, Madan R, Mondal A, et al. Occult phosphaturic mesenchymal tumour of femur cortex causing oncogenic osteomalacia - diagnostic challenges and clinical outcomes. Endokrynol Pol 2018;69:205-10. [Google Scholar | PubMed]
- 9.Uchihashi K, Nishijima-Matsunobu A, Matsuyama A, Yamasaki F, Tanabe T, Uemura T, et al. Phosphaturic mesenchymal tumor, nonphosphaturic variant, causing fatal pulmonary metastasis. Hum Pathol 2013;44:2614-8. [Google Scholar | PubMed]
- 10.Yoshioka K, Nagata R, Ueda M, Yamaguchi T, Konishi Y, Hosoi M, et al. Phosphaturic mesenchymal tumor with symptoms related to osteomalacia that appeared one year after tumorectomy. Intern Med 2006;45:1157-60. [Google Scholar | PubMed]
- 11.Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: An analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol 2004;28:1-30. [Google Scholar | PubMed]






Related Articles in Journal of Orthopaedic Case Reports
 February 10, 2024 Phosphaturic Mesenchymal Tumor in the Proximal Femur Presenting as Tumor-induced Osteomalacia: A Case Report and Literature Review
February 10, 2024 Phosphaturic Mesenchymal Tumor in the Proximal Femur Presenting as Tumor-induced Osteomalacia: A Case Report and Literature Review March 10, 2021 A Case of Tumor-Induced Osteomalacia: Finding the Culprit Acetabular Tumor and Successful Resection with a Novel Hip Joint-Preserving Surgery
March 10, 2021 A Case of Tumor-Induced Osteomalacia: Finding the Culprit Acetabular Tumor and Successful Resection with a Novel Hip Joint-Preserving Surgery January 10, 2020 Osteomalacia and Insufficiency Fractures Secondary to Intravenous Iron Therapy: A Case Report
January 10, 2020 Osteomalacia and Insufficiency Fractures Secondary to Intravenous Iron Therapy: A Case Report November 10, 2022 Phosphaturic Mesenchymal Tumor-induced Osteomalacia Masquerading as an Orthopedic Polytrauma Patient: A Case Report and Review of the Literature
November 10, 2022 Phosphaturic Mesenchymal Tumor-induced Osteomalacia Masquerading as an Orthopedic Polytrauma Patient: A Case Report and Review of the Literature