
Normophosphatemic tumoral calcinosis with ophthalmic involvement.
Dr. Shibanand Buddhia, Department of Orthopaedics, Veer Surendra Sai Institute of Medical Sciences and Research, Burla, Sambalpur, Odisha, India. E-mail: buddhiashibanand@gmail.com
Abstract
Introduction: Tumoral calcinosis (TC) is a rare disorder characterized by periarticular soft-tissue deposition of calcium around large joints.
Case Report: We report a recurrent familial case of a 63-year-old Indian female with progressive painless discharging mass over the left proximal thigh associated with extensive ophthalmological involvement in the form of perilimbal calcific deposits, angioid streak, pigment epithelial defects, and choroidal neovascular membrane in both eyes. Hematological parameters revealed normophosphatemic. Radiological investigation suggested periarticular calcific deposits with sedimentation sign, and the diagnosis was later confirmed on histopathological examination.
Conclusion: We report the case because of its rarity and suggest thorough ophthalmological and genetic evaluation in each and every patient of TC.
Keywords: Recurrent familial, normophosphatemic, tumoral calcinosis, ophthalmological manifestation.
Tumoral calcinosis (TC) is a rare, benign condition characterized by deposits of calcific material in periarticular soft tissues [1]. It most commonly involves the larger joints such as the hip and shoulder, but also has been reported in joints such as the elbow, wrist, knee, scalp, larynx, spine, and sacrum [2]. This condition mostly occurs in adolescents and young adults, but familial forms affecting the infants are also described [3]. These lesions are slowly growing and progress over the years and may be associated with ulcerations of the overlying skin [4]. In 1899, Duret reported two such cases in two siblings and named it as endothelium calcifie [5]. In 1935, Teutschlander labeled it as lipocalcino granulomatosis or Teutschlender disease. In 1943, the name was revised to TC by Inclan et al. [6]. The etiology of TC is inconclusive despite the proposal of many theories [4]. On the basis of underlying etiology, the disease can be primary or secondary to other conditions. Depending upon the phosphate level, primary TC can be normophosphatemic or hyperphosphatemic. The secondary TC is usually secondary to conditions such as chronic kidney diseases, hyperparathyroidism, and hypervitaminosis D [7]. In this article, we report a primary recurrent normophosphatemic familial variety of TC in a female patient.
A 63-year-old female patient with no comorbidities presented to our department with a history of progressive swelling over the left upper thigh with discharge of whitish chalky material from the ulceration over it (Fig. 1a).
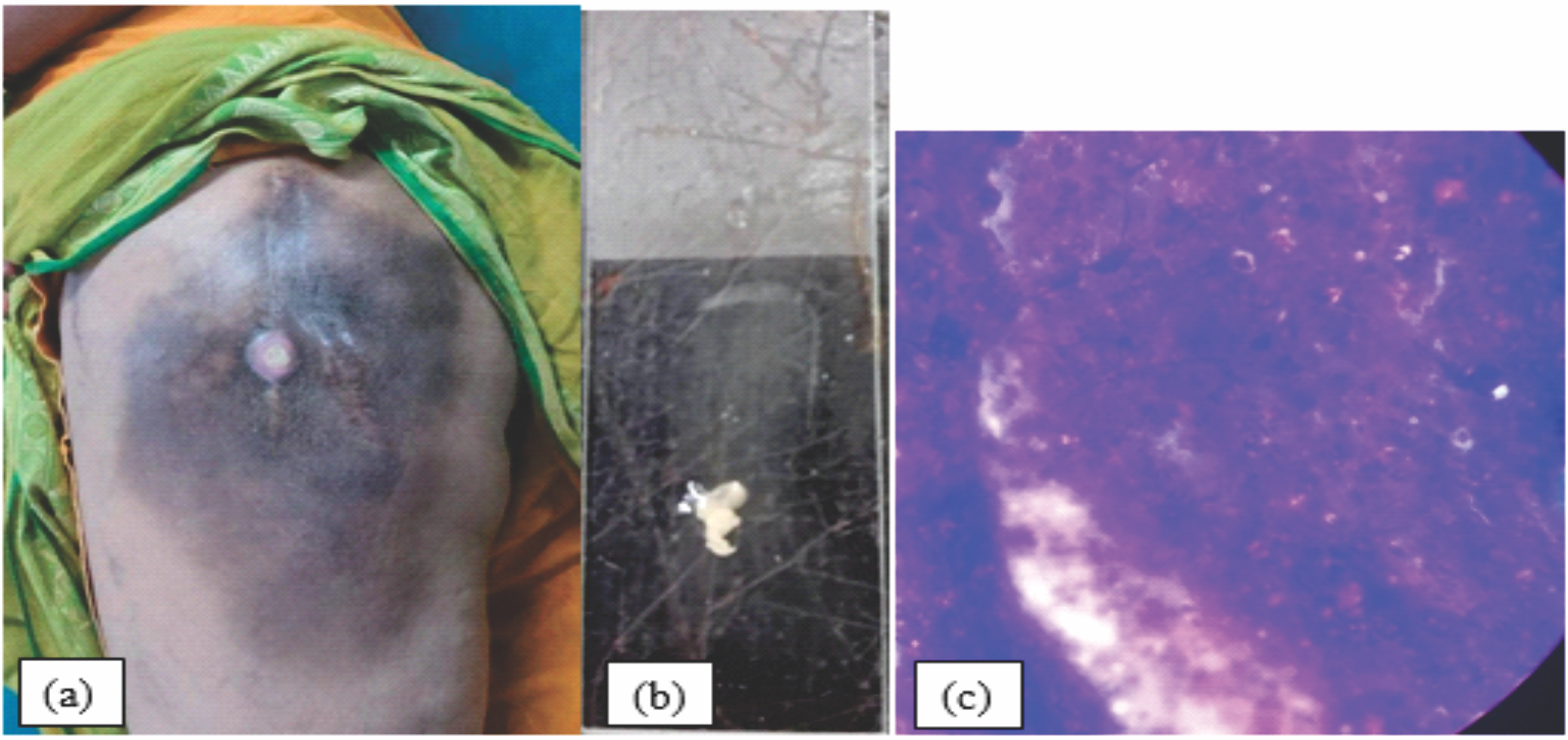
Figure 1: [a]: Clinical picture of the mass with ulceration and hyperpigmentation, [b]: Chalky whitish fluidic discharge from the ulceration. [c] Histological examination of discharge showing a fibrous connective tissue with amorphous calcified material boarded by a florid proliferation of macrophages and multinucleated osteoclast- like giant cells.
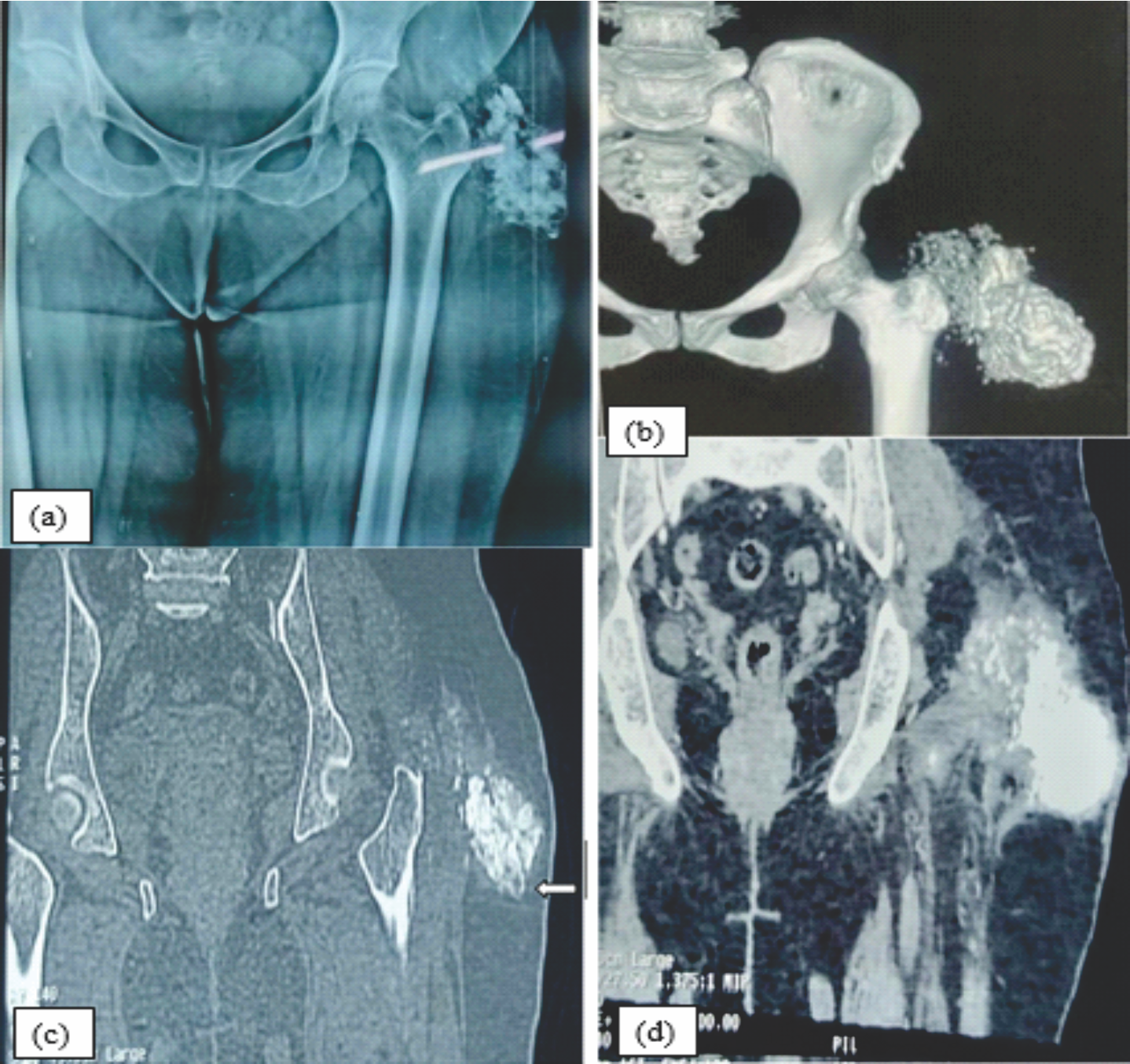
Figure 2: Fig 2[a]: X-ray picture showing a multiobulated periarticular calcified mass around the left hip joint. [b]: CT scan depicting the calcified mass around left hip joint. [c & d]: The coronal CT scan (Bone window & soft tissue window) shows left periarticular multilocular calcified mass with few fluid calcium level (suggestive of sedimentation sign) involving left gluteus maximus muscles with soft tissue extensions around the hips & the upper femur.
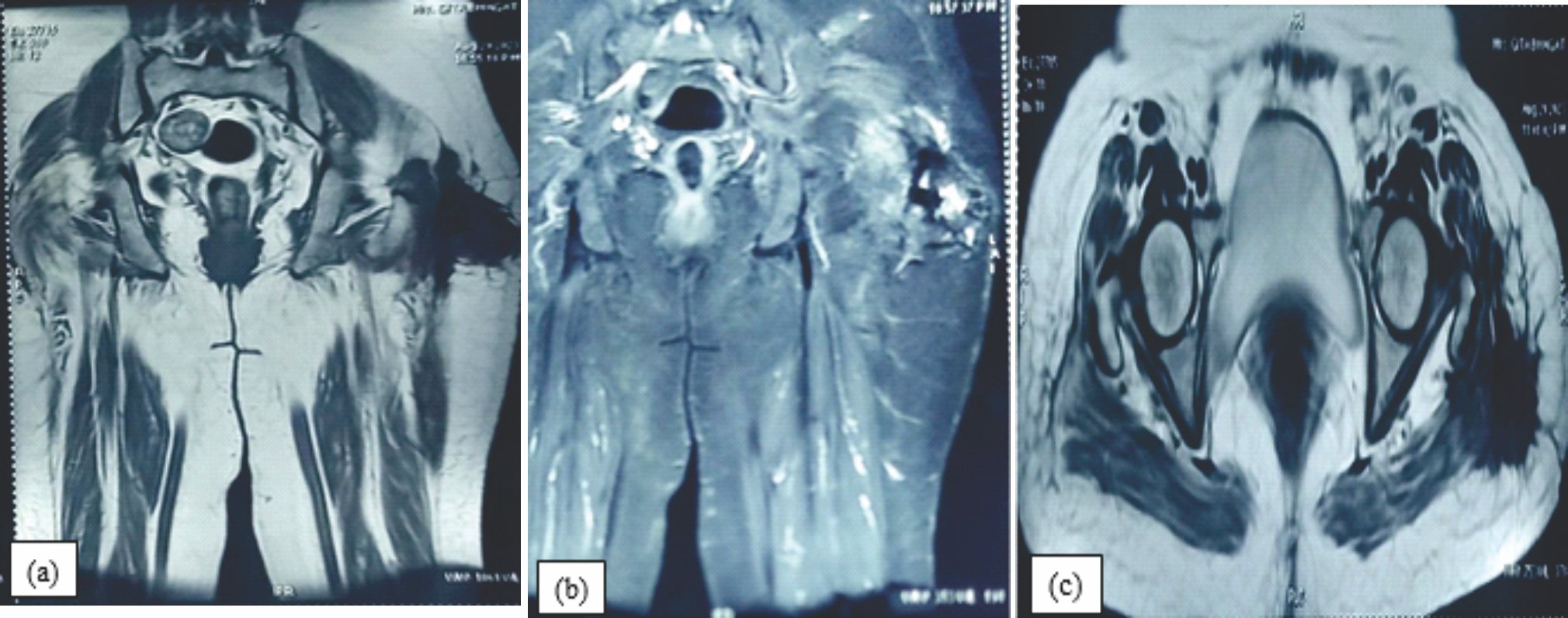
Figure 3: (a, b &c): Coronal T1, STIR & axial T2, images of the hip reveal a periarticular , multilobulated mass with mild internal T2 hyperintensity .The mass is well demarcated, lobulated and predominantly low signal on T2 weighted sequences suggestive of calcification noted in the left side.
Contrast-enhanced computed tomography (CT) of the pelvis revealed an ill-defined, multilobulated amorphous and heterogenous calcified lesion in the subcutaneous plane in the left gluteal region into the left gluteus maximus with minimal involvement of gluteus medius measuring 7.1 × 7.5 × 8.9 cm (Fig. 2b). The coronal CT scan (Bone window and soft tissue window) shows left periarticular multilocular calcified mass with few fluid calcium level suggestive of sedimentation sign (Fig. 2c and d).
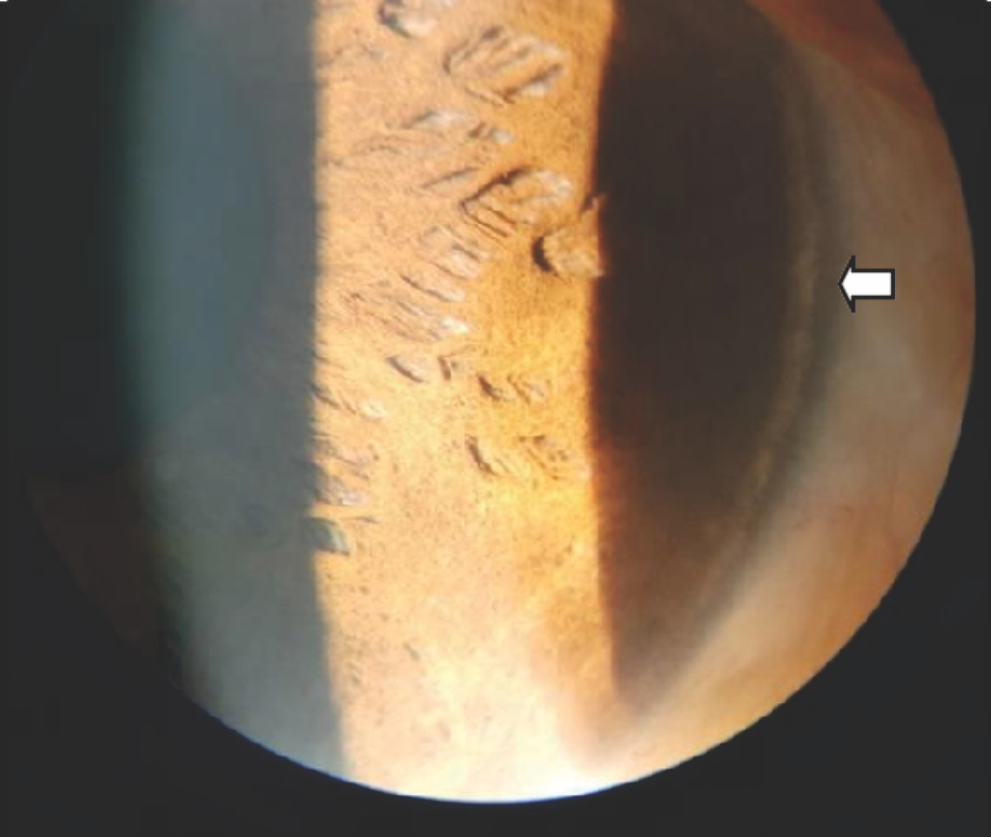
Figure 4: Arrow head showing perilimbal calcific deposit.
On post-contrast images, mild enhancement was seen in the non-calcified soft tissue, and there was no involvement of the bony structure. Magnetic resonance imaging (MRI) revealed a subcutaneous lobulated mass over the left greater trochanteric region of the femur, posterolaterally with fixity to the underlying deep fascia and gluteus maximus. Overlying skin showed focal thickening and scar formation (Fig. 3a, b, c).
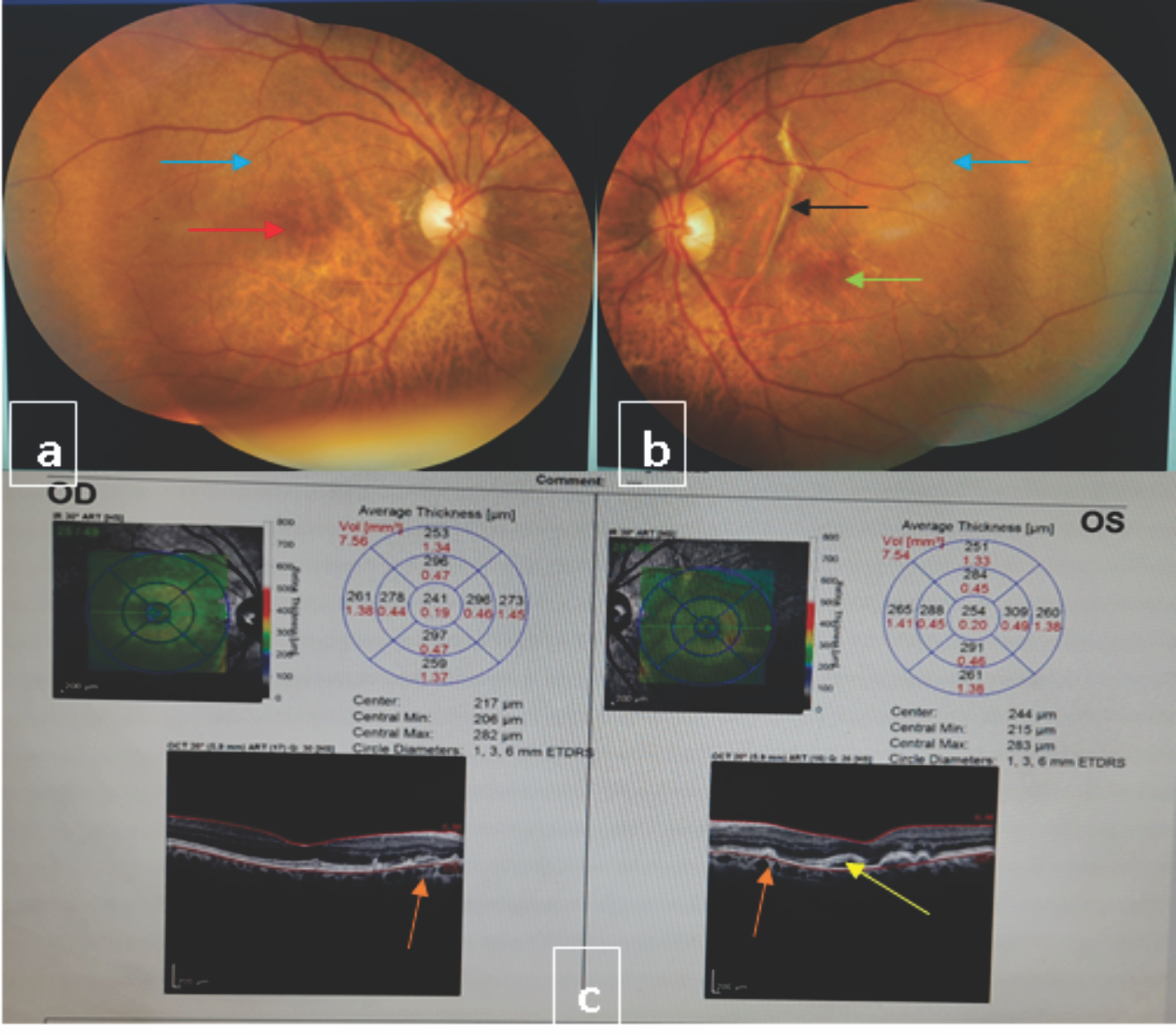
Figure 5: Fundus and Optical Coherence Tomography (OCT) picture of right and left eye (a) Red arrow showing pigment epithelial defects and blue arrow showing calcific deposits in right eye fundus (b) Black arrow shows angioid streak, green arrow representing pigment epithelial defects and blue arrow pointing dotted spot of calcifying deposits in left eye fundus. (c) OCT picture both eye showing multiple pigmented epithelial defects (yellow arrow) and choroidal neovascular membranes (Orange arrow).
Laboratory investigations showed normal phosphate value, without any hematologic value alterations of 1,25-dihydroxyvitamin D or parathyroid hormone. Renal function was normal. Histological examination of the discharge taken from the sinus showed a fibrous connective tissue with amorphous calcified material boarded by a florid proliferation of macrophages and multinucleated osteoclast-like giant cells (Fig. 1c).
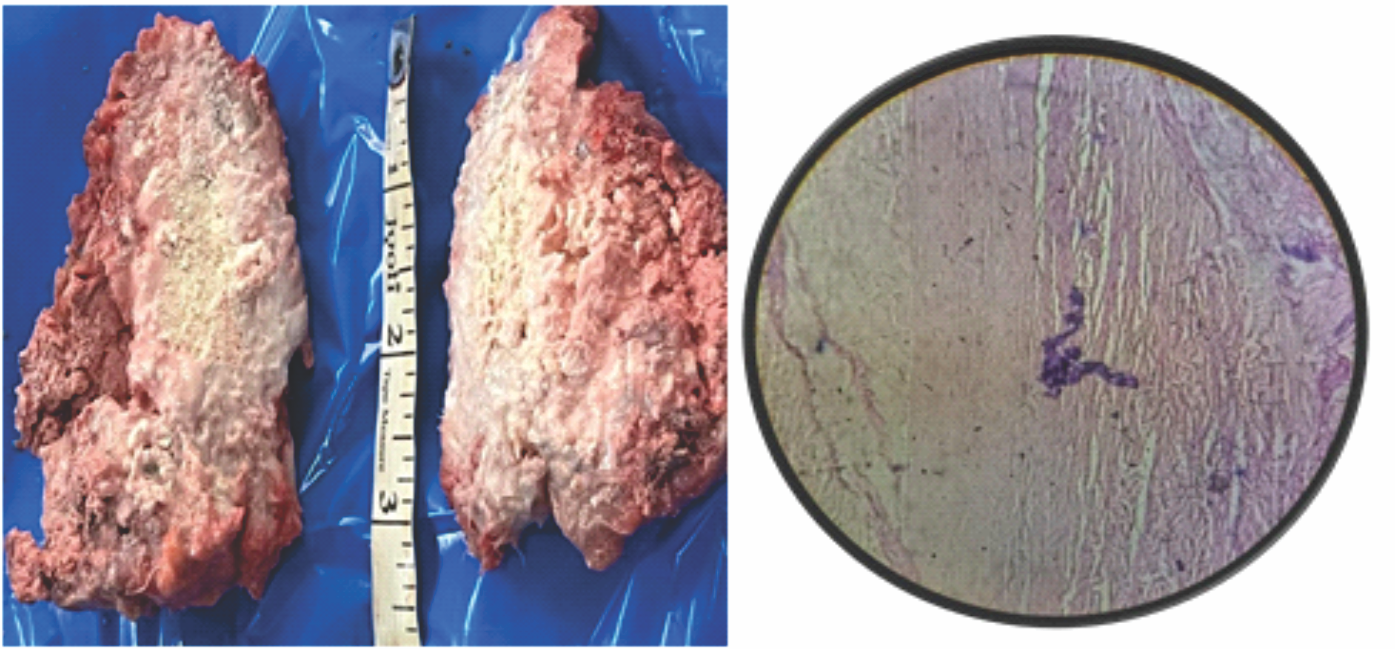
Figure 6: [a]: Gross appearance of the en bloc specimen, [b]: Microscopic section revealing calcification and fibrosis.
Based on the underlying etiology, Smack et al. [7] divided TC into primary or secondary to other conditions. In primary normophosphatemic types, both serum calcium and phosphorus levels are the same without any other hematological abnormalities. It usually presents before the 2nd decade of life, and this variant is familial, involving the gene encoding the SAMD9 protein [7]. In our reported case, repeated hematological investigation showed normophosphatemia, and the patient presented her first episode in the 2nd decade, for which she had undergone surgery somewhere else. Familial form is also confirmed, as one of her sisters is also affected with a similar presentation. The second group is the primary hyperphosphatemic type, usually presenting during the first and second decades of life. This group of patients has a genetic predisposition with reduced urinary phosphate excretion caused by autosomal recessive mutations in GALNT3 and KLOTHO that cause inactivation of FGF23, a phosphaturic hormone. The third group encompasses secondary TC connected with chronic renal failure, primary hyperparathyroidism, hypervitaminosis D, sarcoidosis, etc. [7]. Despite its different etiopathogenesis, the clinical and histopathological features are almost similar to those of our reported case. Joints most commonly affected include the hip, elbow, shoulder, foot, and wrist [4,8]. Apart from this, small joints of the hand, such as proximal and distal interphalangeal joints, are also involved [9]. These lesions are slow-growing and progress over the years. Lesions may be associated with ulceration of the overlying skin with characteristic chalky white liquid discharge material [10]. In children, bone marrow sclerosis and periosteal reactions have also been reported, suggesting its association with chronic recurrent multifocal osteomyelitis [3]. Ophthalmological findings include scleral calcification, limbal calcific deposits, angioid streaks, and subretinal neovascular membrane [11,12]. Angioid streak in the retina may be related to GALNT3 or FGF23 gene mutation [13]. However, we also noted a similar finding, although the above case represents familial normophosphatemic TC. In some cases, calcific deposits in the eyelid and conjunctiva are also seen. There are also possibilities of dental involvement in the form of pulp cavity calcification [14]. Preoperatively, TC is diagnosed by various radiological techniques. Findings of amorphous, multiloculated, and cystic calcifications in a periarticular location on plain radiographs are suggestive of TC [8]. This well-circumscribed, multilobular radiological appearance is described as a cobblestone or chicken wire appearance. CT serves as a guide for surgical excision. It detects the periarticular calcified mass with sedimentation sign (cystic loculi with fluid levels caused by calcium layering) [15]. In MRI, the lesion usually has non-homogeneous diffuse low signal intensity in T1-weighted sequences and alternating signal patterns on T2-weighted images with low signal intensity and cystic components with fluid levels (MRI sedimentation sign) [15]. Complete surgical excision has been classically described for the primary type, although it is associated with high recurrence, failure of complete excision, persistence of etiology, high complication rate, and secondary infection [16]. Even wider surgical excision, keeping an adequate healthy margin, has also not been proven to prevent recurrence [17]. Recurrence is managed by repeated excision. In hyperphosphatemic variants, in order to prevent recurrence, medical managements such as dietary phosphate restriction, use of phosphate binders, acetazolamide, etc., are added postoperatively [18].
TC is a rare diagnosis with poorly defined etiologies. We report an unusual recurrence with a metabolically active lesion in an adult female of familial variety following a long period of quiescence. Considering the rarity of the case, difficulty in diagnosis, and ophthalmological findings, the present case has been reported. Clinical features, careful evaluation of imaging findings, and biochemical profile help in reaching the diagnosis. Biopsy is rarely indicated, only in suspicion of malignancy. Wide surgical excision with or without medical treatment can prevent recurrence.
Although TC is a rare entity, a high index of suspicion is needed to rule it out in patients presenting with periarticular calcific deposits. Once a diagnosis is made, all patients should be screened for possible ophthalmological involvement.
References
- 1. Bassou DA, El Kharras A, Darbi A. Normophosphatemic tumoral calcinosis: A report of case. Eurorad Musculoskelet Syst 2008;3:6388. [Google Scholar] [PubMed]
- 2. Vaghasiya P, Shetty L, Shetty MS, Kumar MA. Tumoral calcinosis: Case report and review. J Health Allied Sci NU 2020;10:90-6. [Google Scholar] [PubMed]
- 3. Kadlec J, Hucin B, Tlaskal T, Westaby S. Aggressive tumoral calcinosis in an infant thoracotomy scar. Interact Cardiovasc Thorac Surg 2010;11:864-5. [Google Scholar] [PubMed]
- 4. Altaf J, Rashid T, Husain M, Arif M, Ahmad M. Tumoral calcinosis, a diagnostic dilemma: A case report. Int J Adv Med 2020;7:1286-9. [Google Scholar] [PubMed]
- 5. Duret MH. Tumeurs et singuliere des bourses (endotheiomes peut etre d’origine paraasitair). Bull Mem Soc Anat Paris 1899;74:275-7. [Google Scholar] [PubMed]
- 6. Inclan A, Leon PP, Camejo M. Tumoral calcinosis. J Am Med Assoc 1943;121:490-5. [Google Scholar] [PubMed]
- 7. Smack D, Norton SA, Fitzpatrick JE. Proposal for a pathogenesis-based classification of tumoral calcinosis. Int J Dermatol 1996;35:265-71. [Google Scholar] [PubMed]
- 8. Olsen KM, Chew FS. Tumoral calcinosis: Pearls, polemics, and alternative possibilities. Radiographics 2006;26:871-85. [Google Scholar] [PubMed]
- 9. Banshelkikar SN, Argekar H, Bhoir A. Idiopathic tumoral calcinosis with unusual presentation-case report with review of literature. J Orthop Case Rep 2014;4:59-62. [Google Scholar] [PubMed]
- 10. Fathi I, Sakr M. Review of tumoral calcinosis: A rare clinico-pathological entity. World J Clin Cases 2014;2:409-14. [Google Scholar] [PubMed]
- 11. McGrath E, Harney F, Kinsella F. An ocular presentation of familial tumoral calcinosis. BMJ Case Rep 2010;2010: bcr0520103044. doi: 10.1136/bcr.05.2010.3044 [Google Scholar] [PubMed] [CrossRef]
- 12. Ghanchi F, Ramsay A, Coupland S, Barr D, Lee WR. Ocular tumoral calcinosis. A clinicopathologic study. Arch Ophthalmol 1996;114:341-5. [Google Scholar] [PubMed]
- 13. Bhattacharjee H, Bhattacharjee K, Yambem DK. Ocular involvement in tumoral calcinosis. Indian J Ophthalmol 2014;62:884-7. [Google Scholar] [PubMed]
- 14. Tiwari V, Zahra F. Hyperphosphatemic Tumoral Calcinosis. Vol. 23. Treasure Island, FL: StatPearls Publishing; 2023. p. 1-9. [Google Scholar] [PubMed]
- 15. Martinez S, Vogler JB 3rd, Harrelson JM, Lyles KW. Imaging of tumoral calcinosis: New observations. Radiology 1990;174:215-22. [Google Scholar] [PubMed]
- 16. King JJ, Brennan KB, Crawford EA, Fox EJ, Ogilvie CM. Surgical complications associated with extensive tumoral calcinosis. Am J Orthop (Belle Mead NJ) 2011;40:247-52. [Google Scholar] [PubMed]
- 17. Fathi I, Sakr M, Hamza Y, Nabawi A. Recurrent tumoral calcinosis: Report of two cases. J Case Rep 2016;6:94-9. [Google Scholar] [PubMed]
- 18. Kirk TS, Simon MA. Tumoral calcinosis. Report of a case with successful medical management. J Bone Joint Surg Am 1981;63:1167-9. [Google Scholar] [PubMed]




Related Articles in Journal of Orthopaedic Case Reports
 January 1, 2025 Pre-patellar Tumoral Calcinosis of Knee with Intra-articular Extension: An Index Case Study
January 1, 2025 Pre-patellar Tumoral Calcinosis of Knee with Intra-articular Extension: An Index Case Study May 10, 2022 Reviewers Acknowledgement & Photo Gallery May 2022
May 10, 2022 Reviewers Acknowledgement & Photo Gallery May 2022 January 10, 2024 Primary Hyperparathyroidism Mimicking Skeletal Metastasis – A Diagnostic Dilemma
January 10, 2024 Primary Hyperparathyroidism Mimicking Skeletal Metastasis – A Diagnostic Dilemma December 1, 2025 Tibialization of Fibula in an Implant Failure Non-Union Poliotic Limb: A Case Report
December 1, 2025 Tibialization of Fibula in an Implant Failure Non-Union Poliotic Limb: A Case Report