[box type=”bio”] Learning Point of the Article: [/box]
Hyperflexion and rotation trauma of the knee joint result rather in bony avulsions than in ligamentous lesions in paediatric patients.
Case Report | Volume 8 | Issue 5 | JOCR September – October 2018 | Page 36-39| Melih Civan, Fuat Bilgili, Ayse Kilic, ZeyraOya Uyguner, Guven Toksoy. DOI: 10.13107/jocr.2250-0685.1200
Authors: Melih Civan[1], Fuat Bilgili[1], Ayse Kilic[2], ZeyraOya Uyguner[3], Guven Toksoy[3]
[1]Department of Orthopedics and Traumatology, Istanbul University, Istanbul School of Medicine, Sehremini, Fatih, Istanbul, Turkey.
[2]Department of General Pediatrics, Istanbul University, Istanbul School of Medicine, Sehremini, Fatih, Istanbul, Turkey.
[3]Department of Medical Genetics, Istanbul University, Istanbul School of Medicine, Sehremini, Fatih, Istanbul, Turkey.
Address of Correspondence:
Dr. Melih Civan,
Department of Orthopedics and Traumatology, Istanbul University, Istanbul School of Medicine, Sehremini, Fatih, Istanbul, Turkey.
E-mail: melihcivan@gmail.com
Abstract
Introduction: Fibrodysplasia ossificans progressiva previously known as myositis ossificans progressiva is a rare connective tissue disorder with autosomal dominant genetic inheritance. Patients develop heterotrophic ossification starting with the first decade of life. Diagnosis is extremely difficult until ossifications are visible.
Case Report: We report a case of fibrodysplasia ossificans progressiva in a 5-year-old boy who has characteristic extracapsular joint movement limitation with bilateral great toe malformation. Before clinical suspicion and genetic confirmation, the patient had undergone various medical tests including biopsy. The patient was diagnosed by the help of characteristic great toe malformations with the help of X-ray taken after ossification signs revealed.
Conclusion: Fibrodysplasia ossificans progressiva is an unforgiving disease. Late diagnosis can lead the physicians to perform additional invasive test and restrains patients to avoid the exposure of more daily trauma. Although there is no treatment for the disease in current literature, we believe with the characteristic features, it could be diagnosed in short notice and managed properly.
Keywords: Ossification, Fibrodysplasia, Diagnosis, Hallux valgus.
Introduction
Fibrodysplasia ossificans progressiva (FOP, Online Mendelian Inheritance in Man [OMIM] 135100) is a very rare, inherited connective tissue disorder. Patients develop heterotrophic ossification in tissues such as muscles, tendons, ligament, fascia, and connective tissue. Ossification usually starts at the first decade of life. In 2006, ACVR1 gene, which is primary responsible for this disease has been described [1]. Although a heterozygote point mutation (c.617G>A; p.Arg206His) is primary responsible for the disease, there are also other mutations described in the literature such as c.774G>T; p;Arg258Ser [2,3]. It is crucial to make an early diagnosis for these patients, yet it is difficult until ossifications become radiologically visible. Misdiagnosis rates could be up to 90%. The disease should be distinguished mainly from Progressive Osseous Heteroplasia (OMIM 166350), Albright hereditary osteodystrophy, and McCune-Albright syndrome (OMIM 103580). Furthermore, it is usual to investigate patients for aggressive juvenile fibromatosis, lymphedema, soft tissue sarcomas, desmoid tumors, and muscular dystrophies before diagnosis of FOP [4]. We will report a 5-year-old male FOP patient diagnosed after long medical investigation period.
Case Report
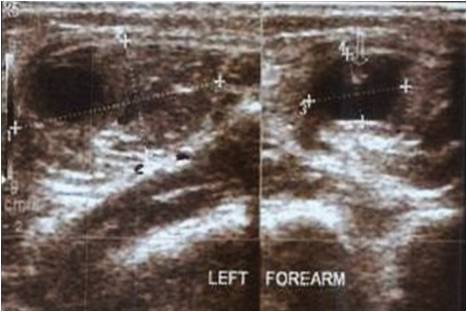
A 5-year old boy had a 2-weeks history of a fall at school. At the time of presentation, physical examination revealed a diffuse elastic soft mass behind his neck and below his right scapula. His shoulder abduction was limited to 90° bilaterally. He had also limited cervical movements in lateral rotation and flexion. He was unable to put his clothes on by himself. Ultrasound, peripheral blood analysis, and biochemistry parameters were normal. There were no malignancies detected. X-ray and magnetic resonance imaging (MRI) revealed fusion anomalies at the posterior elements of cervical and upper thoracic spine (Fig. 1,2).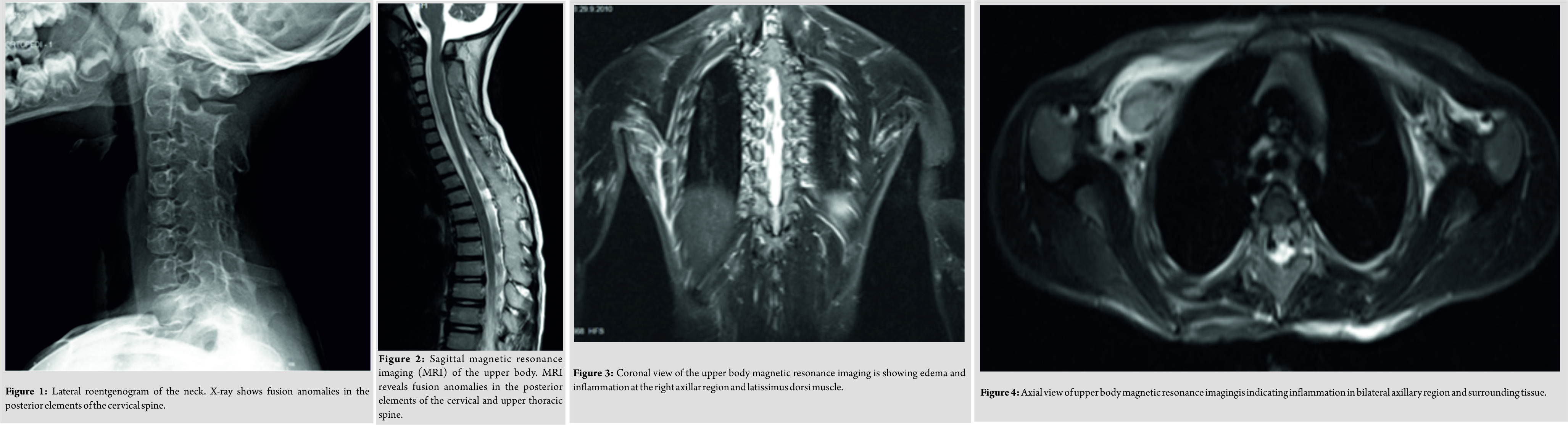 All immunological and tumor markers were negative. Lymphoscintigraphywas normal. HLAB27 was the only positive immunologic parameter. In 3 weeks, significant loss of abduction in shoulder joints was noted bilaterally. MRI revealed enhanced edema at bilateral axillary region (Fig. 3, 4).
All immunological and tumor markers were negative. Lymphoscintigraphywas normal. HLAB27 was the only positive immunologic parameter. In 3 weeks, significant loss of abduction in shoulder joints was noted bilaterally. MRI revealed enhanced edema at bilateral axillary region (Fig. 3, 4).
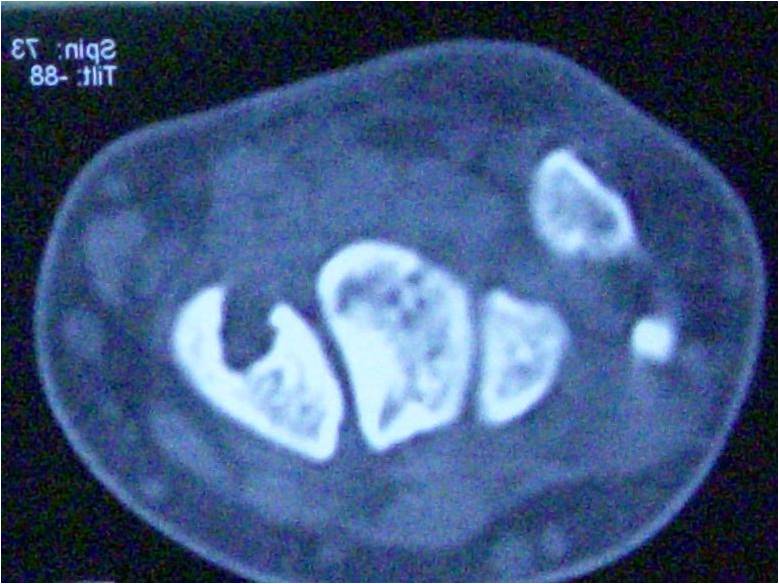
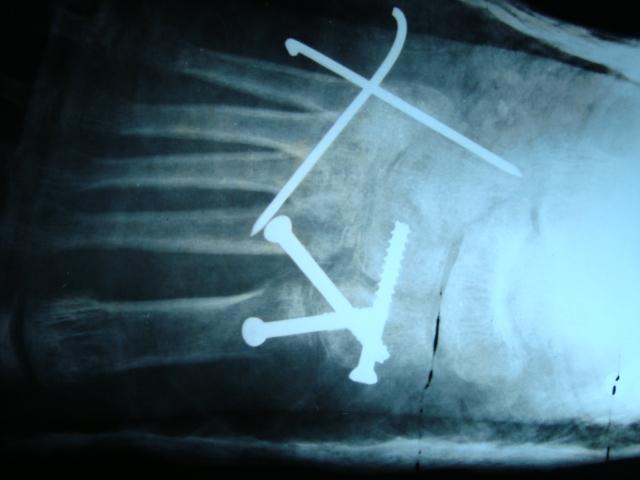
Biopsy showed inflammatory changes with increased fibroblastic activity. There were no signs of muscular diseases. At 5 weeks after initial presentation, axillar heterotrophic ossification was detected in X-ray (Fig. 5, 6) with concomitant characteristic bilateral great toe malformations and hallux valgus (Fig. 7). Bilateral short broad femoral necks supported our diagnosis (Fig. 8). There were swellings and palpable mass at the medial side of the left scapula and lumbar paraspinal muscle area where the biopsy was performed (Fig. 9).At the end, genetic analysis showed heterozygous mutation c.617G>A p.Arg206His and confirmed clinical and radiological diagnosis of FOP.
Discussion
Fibrodysplasia ossificans progressiva is an autosomal dominant inherited disease diagnosed in the first decade of life with a prevalence of one case in 2 million individuals. There is no ethnic, racial, or geographic predisposition described [1, 2, 3, 4]. The disease could be also seen with spontaneous mutations. FOP-related ACVR1 gene is positioned on the 2q23-24 chromosome and codes bone morphogenetic protein type-1 receptor [1]. Trauma, viral infections, dental procedures, surgery, or vaccinations could directly initiate the disease or could trigger new ossifications [5, 6, 7]. These events could also exacerbate the flare-ups after diagnosis [2]. We believe that the triggering event was physical trauma at school for our patient. Before ossification, congenital malformations at great toes or hallux valgus could be an alarming clinical sign for referring patient to the genetic analysis [8]. Yet we were unable to recognize the great toe malformations before ossifications revealed in X-ray. Painful soft tissue swellings seen after onset of the disease could easily be mistaken as tumors. In our case, these swellings were inspected for malignancies. The diaphragm, tongue, extraocular muscles, cardiac, and smooth muscles are not affected in the FOP. Progressive extracapsular ankylosis at the affected joints or neck stiffness could be the initial symptom. Characteristically, cervical movements could be limited by fusion of the facet joints from C2 to C7 [9]. Posterior elements could be large while vertebral bodies are tall and narrow. Other clinical features related to FOP are clinodactyly, short broad femoral neck, proximal medial tibial osteochondromas, ankylosis of the jaw, right-sided heart failure, kyphoscoliosis, lymphedema, and hearing loss [10,11]. Great toe malformation is pathognomonic; however, it is not present in all patients [8]. We have detected bilateral short broad femoral neck and cervical spinal fusion in our patient, and yet we did not relate these findings to any syndrome in the beginning. Late or missed diagnosis can cause multiple invasive procedures such as biopsy or surgery which both are contraindicated and harmful [4]. We performed needle and one open muscle biopsy to the patient and found irregular collagen and fibroblastic proliferation with inflammation similar to the literature [12]. Biochemical evaluations and advanced imagining methods do not contribute to diagnosis. MRI could only reveal inflammation in early stages. Double calcaneal ossification centers in early infancy or calcaneal spurs are promising radiologic features for early diagnosis [13]. The best and only way to make an early diagnosis and prevent higher medical costs is to detect characteristic skeletal features of the disease along with the specific medical history. In fact, after genetic confirmation of the disease, there is not much to do in current knowledge. Steroids and muscle relaxants could be used at the beginning of the flare-up periods. NSAIDs, mast cell inhibitors, or narcotic analgesics could also be used during or after flare-up periods. Children’s daily activities and school education program must be changed for preventing more falls and traumas. Protective gears, house education, and acceptable activity modifications are useful solutions. Immunizations, dental care, anesthesia, and surgeries must be performed in accordance with the established guidelines [14]. With current medical literature, the approximate duration of life is four decades. The second half is usually dependent to the wheelchair. Common causes of death for this disease are thoracic insufficiency syndrome or other pulmonary complications [15].
Conclusion
Today, there is not an actual medical or surgical treatment for FOP patients. However, there are promising studies and clinical trials. We believe that the most beneficial act is to make an early diagnosis and do not harm for the rest of their lives. Multidisciplinary approach should guide the parents to modify their children’s daily activities or school life. Even some of them could be protected better at home education for the new traumatic events. This management can be changed in the future by the great effort of dedicated scientist working on the treatment.
Clinical Message
Early diagnosis prevents harmful invasive procedures and facilitates the further management of the disease.
References
1. Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat2009;30:379-90.
2. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006;38:525-7.
3. Bocciardi R, Bordo D, Di Duca M, Di Rocco M, Ravazzolo R. Mutational analysis of the ACVR1 gene in italian patients affected with fibrodysplasia ossificans progressiva: Confirmations and advancements. Eur J Hum Genet 2009;17:311-8.
4. Kaplan FS, Shore EM, Connor JM. Fibrodysplasia ossificans progressiva. In: Royce PM, Steinmann B, editors. Connective Tissue and its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2nd ed. New York: John Wiley and Sons; 2002. p. 827-40.
5. Glaser DL, Rocke DM, Kaplan FS. Catastrophic falls in patients who have fibrodysplasia ossificans progressiva. Clin OrthopRelat Res 1998;346:110-6.
6. Scarlett RF, Rocke DM, Kantanie S, Patel JB, Shore EM, Kaplan FS, et al. Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva. Clin OrthopRelat Res 2004;423:275-9.
7. Lanchoney TF, Cohen RB, Rocke DM, Zasloff MA, Kaplan FS. Permanent heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. J Pediatr1995;126:762-4.
8. EresenYazıcıoğlu C, Karatosun V, Kızıldağ S, Ozsoylu D, Kavukçu S. ACVR1 gene mutations in four Turkish patients diagnosed as fibrodysplasia ossificans progressiva. Gene 2013;515:444-6.
9. Schaffer AA, Kaplan FS, Tracy MR, O’Brien ML, Dormans JP, Shore EM, et al. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with klippel-feil syndrome: Clues from the BMP signaling pathway. Spine (Phila PA 1976) 2005;30:1379-85.
10. Kaplan F, Glaser D, Shore E, Deirmengian G, Gupta R, Delai P, et al. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab2005;3:183-8.
11 Morales-Piga A, Bachiller-Corral J, González-Herranz P, Medrano-SanIldelfonso M, Olmedo-Garzón J, Sánchez-Duffhues G, et al. Osteochondromas in fibrodysplasia ossificans progressiva: A widespread trait with a streaking but overlooked appearance when arising at femoral bone end. Rheumatol Int 2015;35:1759-67.
12. Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 2005;116:e654-61.
13. Hasegawa S, Victoria T, Kayserili H, Zackai E, Nishimura G, Haga N, et al. Characteristic calcaneal ossification: An additional early radiographic finding in infants with fibrodysplasia ossificans progressiva. PediatrRadiol2016;46:1568-72.
14. Kaplan FS, Shore EM, Pignolo RJ. The medical managment of fibrodysplasia ossificans progressiva: Current treatment considerations. Clin Proc Intl Clin Consort FOP 2011;4:96-100.
15. Kaplan FS, Zasloff MA, Kitterman JA, Shore EM, Hong CC, Rocke DM, et al. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am 2010;92:686-91.
 |
 |
 |
 |
 |
| Dr. Melih Civan | Dr. Fuat Bilgili | Dr. Ayse Kilic | Dr. ZeyraOya Uyguner | Dr. Guven Toksoy |
| How to Cite This Article: Civan M, Bilgili F, Kilic A, Uyguner Z, Toksoy G. A Case of Fibrodysplasia Ossificans Progressiva in a 5-year-old Boy with all Musculoskeletal Features and Review of the Literature. Journal of Orthopaedic Case Reports 2018 Sep-Oct; 8(5): 36-39. |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com