
Concomitant congenital deformities and intracranial gliomatic lesions in Neurofibromatosis 1 enhance the possibility for the development of Hypophosphatemic Osteomalacia in which the administration of oral calcitriol and phosphate is an effective treatment in order to improve the clinical symptoms of the disease
Dr. Angelos Kaspiris, MD, MSc, PhD Department of Orthopaedic Surgery, University of Patras 26504, Greece. E-mail: angkaspiris@hotmail.com
Abstract
Introduction: Neurofibromatosis Type 1 (Nf1), also termed von Recklinghausen disease, is a rare autosomal dominant genetic disorder accompanied by several osseous and skeletal manifestations. In NF, hypophosphatemia linked to secondary hyperparathyroidism due to Vitamin D deficiency and low calcium intake has been reported as a risk factor for low bone mass density (BMD), but reports of NF1 associated oncogenic hypophosphatemic osteomalacia (HO) are extremely rare.
Case Report: We report a patient with NF1 associated with intracranial low-grade gliomas and congenital renal agenesis suffering from HO. Bone defects and deformities such as generalized bone pains located in feet, ankles and lower limbs, thoracic scoliosis, mild bowing of long bones of lower limbs, stress fractures, and old fractures as well as with altered bone metabolic serum markers were present. After 8 weeks of follow-up, it was observed that the combination of oral administration of phosphate and Vitamin D improved her medical symptoms without significant changes in phosphate levels or BMD.
Conclusion: Although renal agenesis is not correlated with hypophosphatemia, the coexistence of NF1, renal congenital deformities, and low-grade gliomas may contribute to disease severity. Conventional treatment with high doses of oral calcitriol associated with phosphate is efficient to improve the clinical and laboratory symptoms of the disease.
Keywords: Bone pains, scoliosis, bone mass density, stress fractures, vitamin D.
Neurofibromatosis Type 1 (Nf1), also termed von Recklinghausen disease, is a rare autosomal dominant genetic disorder with a worldwide incidence of 1/2500–3000 individuals [1,2]. NF1 is caused by loss-of-function mutations in the NF1 gene that is located on chromosome 17q11.2 encoding an intracellular protein called neurofibromin (NF), which is responsible for the disease [3]. NF is a cytoplasmic protein consisted of 2818 amino acids and is expressed in the central and peripheral nervous system [4] being involved in the regulation of cellular growth and differentiation through two major pathways: First, NF acts as an Ras-GTPase activating protein regulating negatively the Ras downstream signaling and second, NF functions as an activator of the intracellular levels of cyclic adenosine monophosphate (cAMP), resulting in the inhibition of cell proliferation, differentiation, and survival [5]. Therefore, NF acts as a tumor suppressor protein and biallelic inactivation of NF gene results in lack of its expression in the affected cells stimulating tumor formation process [6,7].
Despite the fact that NF1 is an autosomal dominant disorder with 100% penetrance, there is a great variance in clinical presentation with relatively minor contribution of the nature of the NF1 mutation to disease expression. Diagnostic algorithm is based on the criteria of USA National Institute of Health (NIH) [8] and/or the mutation analysis of NF1 gene. Clinically, it is characterized by the presence of café-au-lait spots, intertriginous freckling, Lisch nodules, neurofibromas, optic pathway gliomas, and distinctive bony lesions. Other features include malignant peripheral nerve sheath tumors, neurocognitive defects, epilepsy, and cardiovascular abnormalities [9]. NF1 is also accompanied by several osseous and skeletal manifestations, including macrocephaly, short stature, sphenoid wing dysplasia, scoliosis, congenital pheudarthrosis of the long bones [10,11], and increased fracture risk [12]. Furthermore, various research studies reported that 20–50% of pediatric and adult patients with NF1 presented with reduced bone mineral density (BMD) or osteoporosis [13,14,15]. Reduced levels of Vitamin D and increased concentrations of parathyroid hormone (PTH), calcium, and bone turnover markers, like tartrate resistant acid phosphatase, were detected in the serum of NF patients with low BMD [16,17]. Histological examinations of bone samples that were received from NF patients with low BMD, revealed reduced volume of trabecular volume, increased osteoid volume and raised number of non-differentiated osteoblastic and osteoclastic cells [18].
Hypophosphatemia linked to secondary hyperparathyroidism due to Vitamin D deficiency and low calcium intake has been reported as a risk factor for low BMD in NF patients [13]. However, reports of NF1 associated oncogenic hypophosphatemic osteomalacia (HO) are extremely rare [13]. This article describes a NF1 patient diagnosed with HO associated with intracranial gliomas and congenital renal agenesis.
A 29-year-old active Caucasian female with multiple facial cutaneous nodules was diagnosed with NF1 according the clinical criteria of NIH [8]. She presented in the outpatient’s department of our institute complaining about progressive bone aches of the feet and ankles, lower limbs, and low-back pain for the past 5 years that were accompanied by muscle weakness, unstable gait, inability to weight bear, and frequent falls. She had also experienced right femoral shaft fracture before 1 ½ years treated with intramedullary nailing (Fig. 1a). She did not suffer from any visual or hearing impairment and her medical history did not reveal renal, liver, gastrointestinal, or constitutional symptoms associated with malignancies. In addition, focal or generalized neurological symptoms were not present. Family history affirmed that her mother was diagnosed with NF1 without evidence of bone metabolic and systematic clinical diseases.
Physical examination displayed a female with short stature (1.52 m with body mass index of 19.8 cm/kg2) with small facial neurofibromas located in the temporal, orbital and cheek areas. Few neurofibromas were also observed over her trunk associated with axillary freckles and numerous café-au-lait macules over her back. Neuromuscular and skeletal examinations were unremarkable. Bilateral multiple Lisch nodules were, also, found during ocular examination.
Skeletal survey with plain radiographs demonstrated generalized demineralization of bilateral lower limb bones (Fig. 1a, b, c) as well as signs of moderate periosteal and endosteal reactions with cortical thickening at the posterior proximal metadiaphyseal region of both tibias (Fig. 1a, b, c), consistent with recent healed stress fractures [19]. Stress fractures were also detected in the lower third of the fibula bilaterally (Fig. 1a, b, c). Despite the fact that feet and ankle aches were prominent symptoms, radiographic examination did not show fracture defects. Full spine X-ray in standing position revealed a right thoracic curve of 11 degrees between T5 and T10 vertebrae without any dystrophic signs (Fig. 2). Bone densitometry measures by DEXA showed low BMD. Spinal sBMD was 914 mg/cm2, while lumbar spinal and hip T and Z-scores were −2 and −2.2, respectively. Cranial magnetic resonance imaging (MRI) detected two low-grade gliotic lesions on the right to putamen and globus pallidus without signs of calcification and ossification or ventricle displacement (Fig. 3). CT-scan and ultrasonography (U/S) of the abdomen were normal, while renal U/S revealed solitary kidney with the left renal agenesis (Fig. 4). Application of quantitative renal scintigraphy with technetium-99m dimercaptosuccinic acid (99mTc‑DMSA) did not detect any disturbances in the structural and functional renal integrity.
Laboratory findings were as follows: Serum calcium was 8.9 mg/dL (reference range between 8.5 and 10.5 mg/dL), serum phosphorous was 1.8 mg/dL (normal: 2.5 and 4.5 mg/dL), serum 25-(OH) Vitamin D, and 1,25-(OH)2 Vitamin D3 were 30.2 ng/ml (normal: 30 and 80 ng/ml) and 55 pg/ml (normal: 16 and 56 pg/ml), respectively, serum alkaline phosphatase and plasma osteocalcin were elevated to the level of 480 iu/L (normal: 35 and 150 iu/L) and 110 ng/ml (normal: 11 and 43 iu/L), respectively. Daily urinary excretion of calcium and phosphorous was 80 mg/24 h (normal range: 0–300 mg/24 h) and 400 nmol/24 h (normal range: 10–40 nmol/24 h), while serum levels PTH, urea, and creatinine were measured within normal limits.
The combination of normal levels of 25-hydroxyvitamin D, PTH, serum calcium, and the concomitant presence of low serum and increased urine secretion of phosphate, as well as the association with bone defects, led us to the diagnosis of oncogenic HO. Patient was treated with oral phosphorous supplementation (2.0 g/day) and calcitriol (1 mg/day). Although during the assessment of the patient 8 weeks after initiation of the treatment, radiological findings were not altered significantly, clinical symptoms, such as foot and lower limb aches or weakness, were markedly improved. Laboratory findings, also, demonstrated elevation in the serum calcium (9.6 mg/dL) but slight improvement in the levels of serum phosphorus (2.0 mg/dL), while the plasma levels of osteocalcin and alkaline phosphatase were not significantly altered.
We presented a case of HO in NF1 accompanied by systematic computer-based literature review search with predefined criteria that were performed in the following databases: PubMed (1947 to present) and Web of Science (1900 to present) using MeSH. The research methodology used a combination of the following terms: “Neurofibromatosis and/or von Recklinghausen (All Fields)”, “hypophosphatemia (All Fields)”, and “oncogenic osteomalacia (All Fields)”. Only full-text articles were eligible for inclusion. Articles written in English, case series, and case reports, case-based reviews were, also, selected for the study. Studies written in a language other than English were excluded from the study.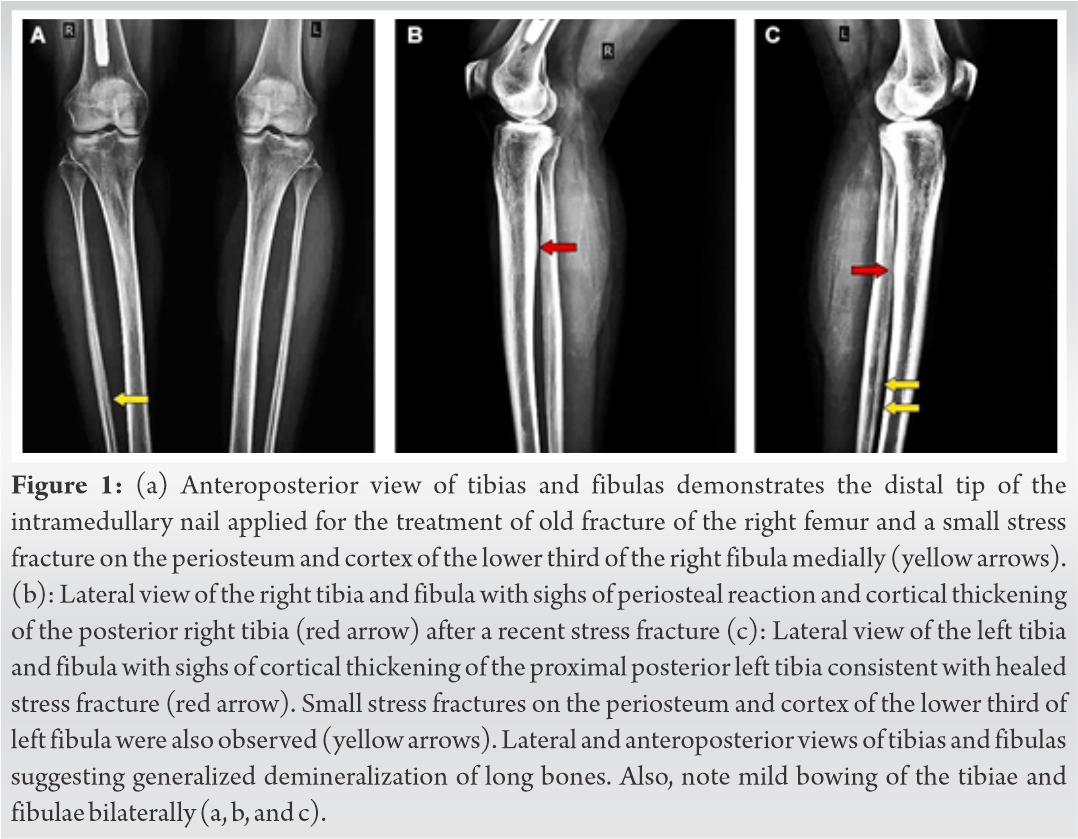
HO secondary to tumors of mesenchymal origin is a rare paraneoplastic syndrome, which is characterized by remarkable low levels of serum and increased concentrations of urine phosphate leading to abnormal bone mineralization [20]. Multiple myelomas, hemangiopericytomas, osteosarcomas, chondroblastomas, chondrofibroid myxomas, malignant fibrous histiocytomas, giant cell tumors, and prostatic cancers are entities that were associated with adult-onset HO [20,21]. NF1 has been implicated with HO very rarely (Table 1) [22,23,23,25,26,27,28,29,30,31,32,33]. Despite the fact, that the association between NF1 and HO was described by Gould in 1918, <50 cases have been referred in the international literature [28]. Moreover, to the best of our knowledge, this is the first report in which HO secondary to NF1 is implicated with low-grade brain gliomas and unilateral renal congenital agenesis.
The combination of clinical manifestations, radiographic, and laboratory examinations contributed in the diagnosis of oncogenic HO. Metabolic data in oncogenic osteomalacia included hypophosphatemia, hyperphosphaturia secondary to reduced proximal renal tubular phosphate reabsorption and low or inappropriate normal levels of serum Vitamin D. Moreover, serum concentrations of calcium and parathormone (PTH) were in normal levels while calcium levels in urine were low [20].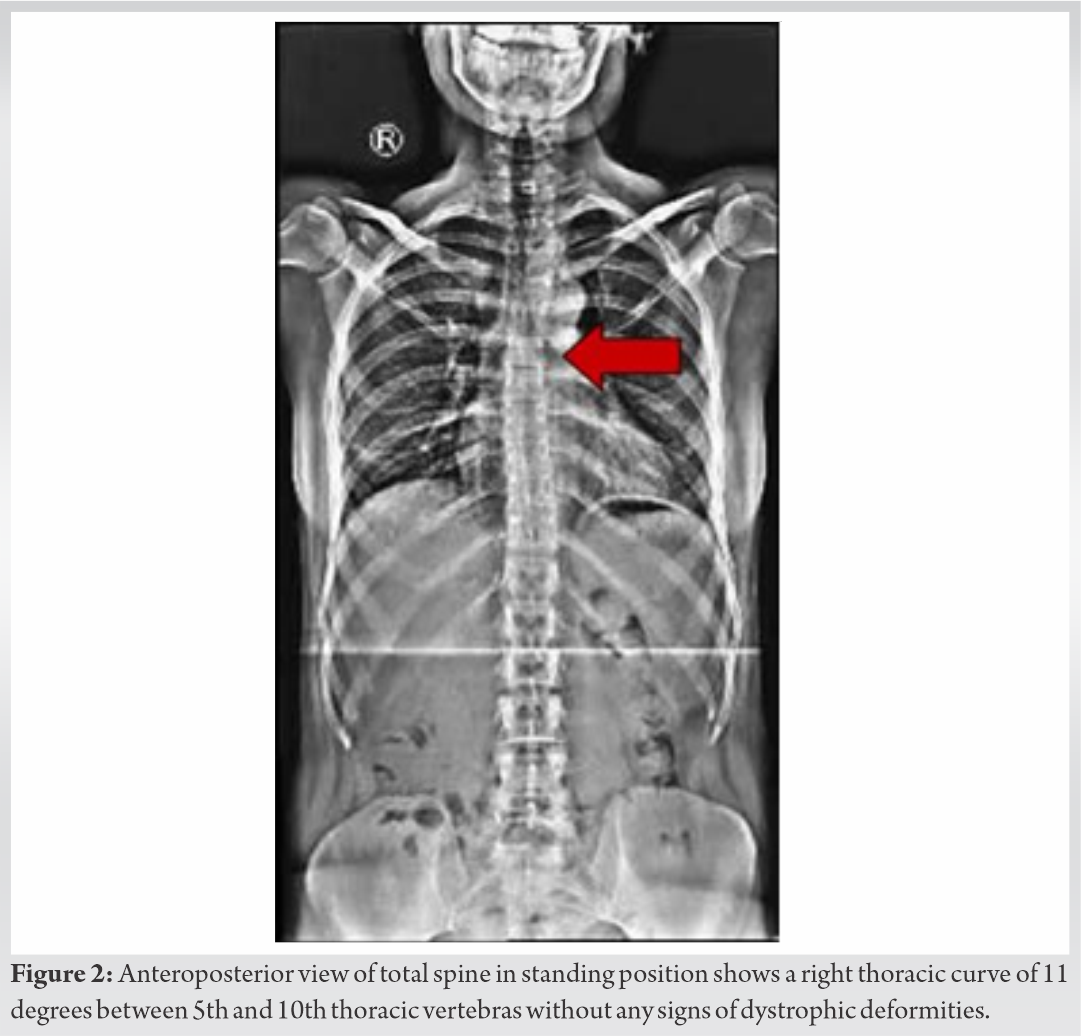
It has been reported that NF1 patients display several skeletal manifestations and deformities [10]. Bone pains, located on the feet and lower limbs as well as muscle weakness, bowing of long bones, pseudofractures, fractures, kyphoscoliosis, and triradiate pelvis, were the most common described defects (Table 1) in NF1 patients with HO [22,23,23,25,26,27,28,29,30,31,32,33]. In our case, generalized bone pains, weakness of the proximal muscles, thoracic scoliosis, mild bowing of long bones of lower limbs, stress fractures, and old healed fractures accompanied by altered bone remodeling serum markers were observed. Interestingly, stress fractures of the distal tibia and fibula were clinically presented with feet and ankles pain, tenderness, and inability to weight bear. Similarly, NF1 patients with HO (Table 1) had low BMD suffering from diffuse osteopenia [27,30] or osteoporosis [25,28,33] and bone demineralization [29,33]. In our report, plain radiographic figures revealed generalized demineralization of the long bones of the lower limbs. In general, the BMD of the patient was low indicating osteopenic defects. This finding was in consistence with the studies of Heerva [13,15] and Lammert [14], in which decreased BMD occurred among patients with NF1 [14] and was accompanied by low bone quality and persistent osteopenia in young patients [15] resulting in generalized bone alteration and osseous dysplasias [14]. Similarly, Jalabert et al. [13] reported that low BMD in NF1 patients was 3.3 times more frequent compared to general population. In the same study, hypophosphatemia was detected in 12 women with NF1 but it was correlated with secondary hyperparathyroidism, and not to oncogenic HO, as it was linked to low calcium intake and Vitamin D deficiency [13].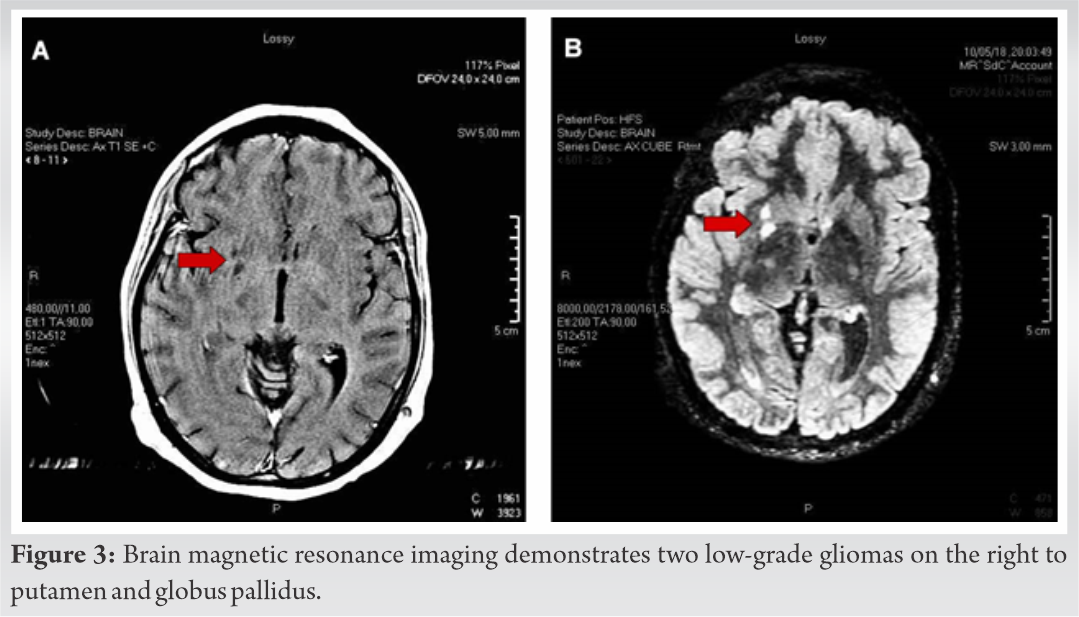
Recently, the implication of fibroblast growth factor 23 or FGF-23 in the pathogenesis of NF1 bone defects with HO was proposed. Specifically, it was suggested that the elevated secretion of FGF-23 from neurofibromin-deficient osteocytes resulted in mineral defects and an osteomalacia – like bone phenotype [33]. Indeed, in conditional knockout for neurofibromin mice model, it was observed that primary osteocytes showed remarkable increase in the expression of FGF-23 in the serum and in the femur [32]. This was linked to abnormal calcium-phosphorus metabolism and to reduced bone formation and mineral apposition rate [34]. In the same study, micro-ct examination, also, demonstrated thinner and porous cortical bones with disorganized osteocyte dendrites that exhibited reduced strength in mechanical forces leading to spontaneous fractures [34]. This was supported by the clinical findings of increased circulating levels of FGF-23 in two patients with NF1 that appeared severe HO [23,25]. A possible explanation was that the increased serum concentration of FGF-23 inhibited renal reabsorption of phosphorus and decreased the production of 1,25–dihydroxyvitamin D leading to increased phosphate wasting and lower levels of phosphorus in the serum [34].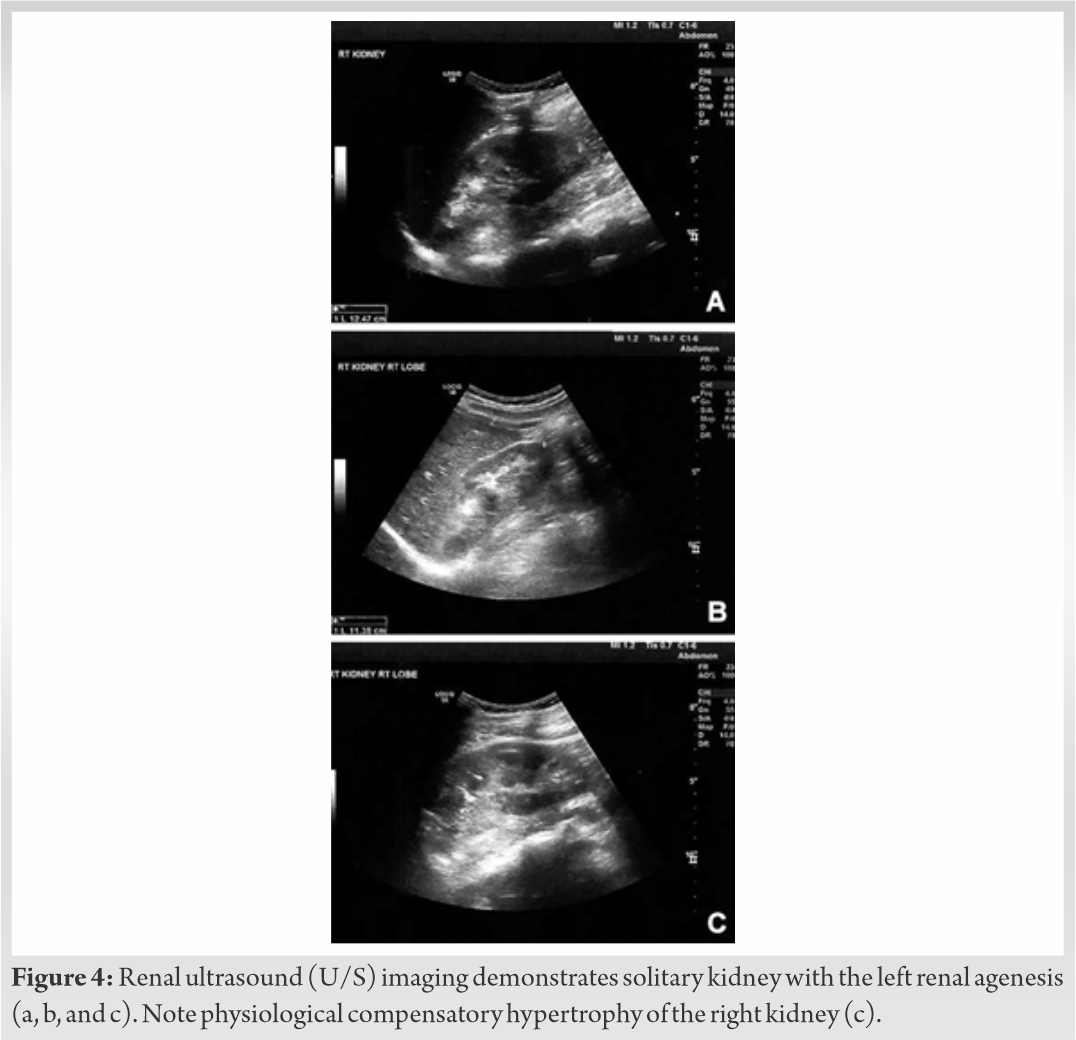
In our report, low-grade gliomatic lesions were also present. Although, association of low-grade gliomas with HO has not been reported in the international literature, we must underline the fact that several angiogenetic growth factors were expressed during gliomas development [35]. The FGF growth factor family played a key role in glioma survivorship. It was reported that increased expression of FGFs (including FGF-23) induced the proliferation, differentiation, and migration of endothelial cells in vitro and the angiogenetic process in vivo [35]. As gliomas showed increased expression of FGF-23, we can speculate that the coexistence of NF1 and low-grade gliomas may enhance the possibility for the development of HO. Unfortunately, our patient did not approve the examination for serum or tissue FGF-23 levels using immunological techniques. However, the above assumption was supported by our finding that 7 years ago, when visible evidences of the gliomatic tumors were not detected, laboratory examinations of the patient were within normal levels, indicating a recent tumor-induced overproduction of FGF23. It must be noted that recent experimental and human immunochemical data suggested that in NF patients, bones and/or concomitant malignancies of mesenchymal origin and not neurofibromas were the primary source of FGF23 overproduction [25].
In our case, NF1 was associated with unilateral renal agenesis that was considered as benign condition. Although it was linked to metabolic abnormalities such as hypercalcuria or hypocitraturia, no correlation between renal agenesis and increased urine loss of phosphorous was observed [36]. However, in animal rat model, renal agenesis was accompanied by increased expression of FGF-23 and, therefore, the contribution of renal agenesis in the HO development cannot be excluded from the study [37]. With a given agenesis of the left kidney, differential diagnosis of conditions of renal phosphate wasting, such as Fanconi syndrome, was deemed necessary. In this report, clinical or laboratory results specifying Fanconi syndrome were not observed. The diagnosis of Fanconi syndrome was based on typical findings of glycosuria (with normal plasma glucose), proteinuria, and phosphaturia along with hypophosphatemia and non-anion gap hyperchloremic metabolic acidosis due to increased renal secretion of bicarbonate. Early serum and urine laboratory examination may reveal the presence of Fanconi syndrome [38].
Although, in our report, NF1 was implicated with intracranial low-grade gliomas and congenital renal disorder, the current therapeutic approach with administration of high doses of oral calcitriol combined with phosphate was efficient and improved the clinical symptoms of the patient. We must highlight the fact that clinical improvement was usually observed during the first 8 weeks of the treatment [22,24,26,27,28,29,30], whereas marked increase in the calcified components of the bone with complete fractures healing and restoration of BMD as well as normalization of the serum markers of bone remodeling such as osteocalcin, alkaline phosphatase, and C-terminal telopeptides were detected after 10–12 months of the treatment [25,27,32]. Similarly, persistent hyperphosphaturia and gradual increase of serum phosphorus were also reported [39]. The role of serum calcium, phosphate, and PTH in regulating FGF23 levels is not clear. In-vitro experimental studies have shown Vitamin D dependent regulation of FGF23 promoter activity, but have failed to identify effects of calcium or phosphate on FGF23 promoter [40]. Moreover, calcitriol was an important modulator of FGF23 production in osteoblasts, potentially regulating FGF23 levels indirectly [40]. Careful monitoring of renal function and possible complications such as hypercalciuria or nephrolithiasis must be considered. However, surgical resection of the tumors or large neurofibromas could be the treatment of choice.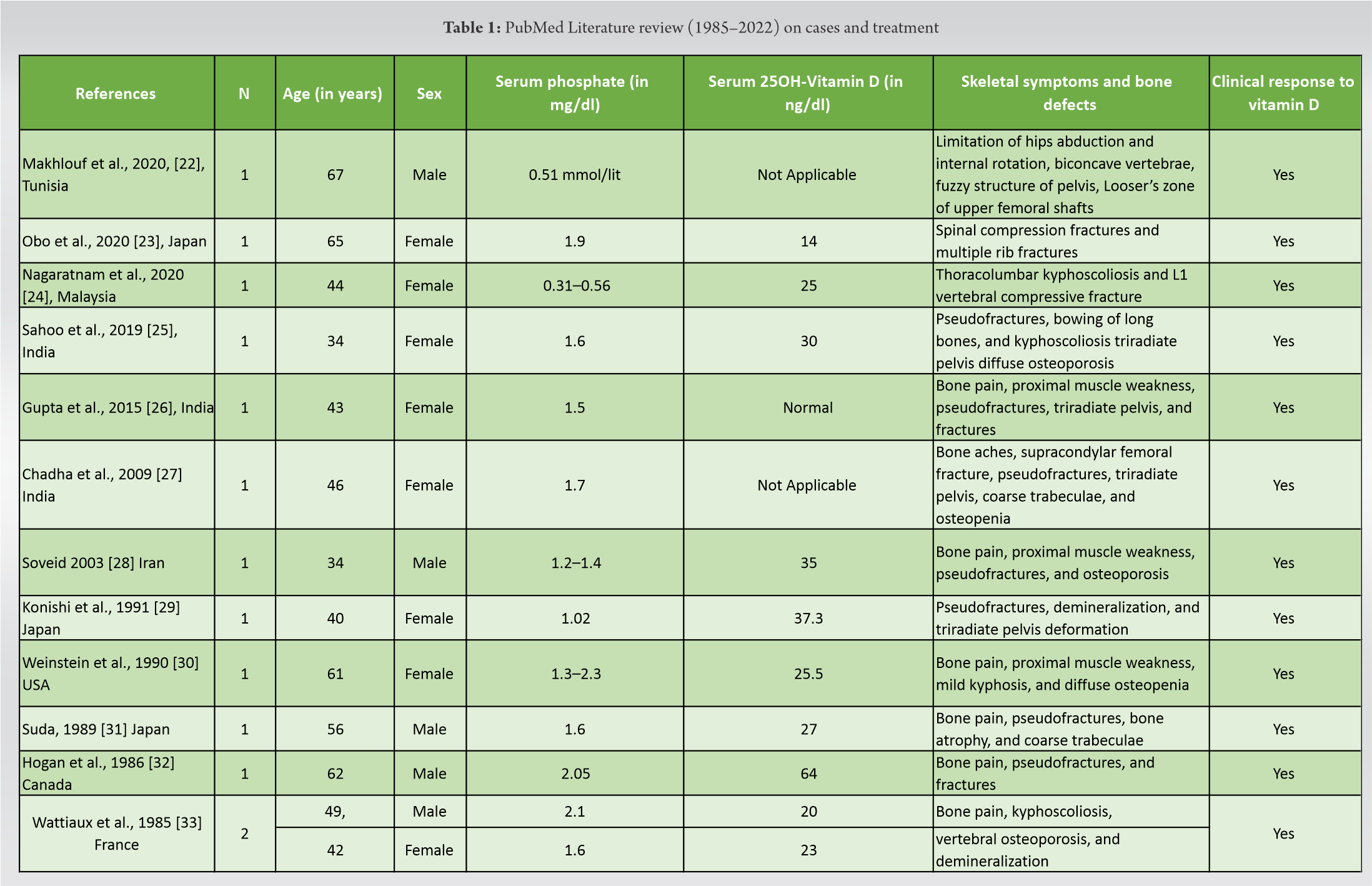
We report a patient with NF1 associated with intracranial low-grade gliomas and congenital renal agenesis suffering from HO. Bone defects and deformities such as generalized bone pains, thoracic scoliosis, mild bowing of long bones of the lower limbs, stress fractures, and old fractures were present. Despite the fact that renal agenesis was not associated with HO, the concomitant presence of NF1 and low-grade gliomas may enhance the possibility of the development of HO. Further studies are deemed necessary to elucidate the exact role of these pathologies in the signaling pathways that result in HO.
Association of Neurofibromatosis 1 with tumors of the central nervous system and/or congenital renal deformities may enhance the possibility of osteopenia and skeletal abnormalities due to hypophosphatemia in young ages. However, conventional treatment with high doses of oral calcitriol combined with phosphate is efficient to improve the clinical and laboratory symptoms of the disease.
References
- 1.LM, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol 2005;141:71-4. [Google Scholar | PubMed]
- 2.Moramarco A, Mallone F, Sacchetti M, Lucchino L, Miraglia E, Roberti V, et al. Hyperpigmented spots at fundus examination: A new ocular sign in neurofibromatosis type I. Orphanet J Rare Dis 2021;16:147. [Google Scholar | PubMed]
- 3.Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med 2010;12:1-11. [Google Scholar | PubMed]
- 4.Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (Nf1): Evidence for modifying genes. Am J Hum Genet 1993;53:305-13. [Google Scholar | PubMed]
- 5.Hannan F, Ho I, Tong JJ, Zhu Y, Nurnberg P, Zhong Y. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum Mol Genet 2006;15:1087-98. [Google Scholar | PubMed]
- 6.Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer 2015;15:290-301. [Google Scholar | PubMed]
- 7.Yap YS, McPherson JR, Ong CK, Rozen SG, Teh BT, Lee AS, et al. The NF1 gene revisited-from bench to bedside. Oncotarget 2014;5:5873-92. [Google Scholar | PubMed]
- 8.Stumpf DA. Neurofibromatosis. Conference statement, National institute of health development conference. Arch Neurol 1988;45:575-8. [Google Scholar | PubMed]
- 9.Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009;123:124-33. [Google Scholar | PubMed]
- 10.Elefteriou F, Kolanczyk M, Schindeler A, Viskochil DH, Hock JM, Schorry EK, et al. Skeletal abnormalities in neurofibromatosis type 1: Approaches to therapeutic options. Am J Med Genet A 2009;149A:2327-38. [Google Scholar | PubMed]
- 11.Stevenson DA, Little D, Armstrong L, Crawford AH, Eastwood D, Friedman JM, et al. Approaches to treating NF1 tibial pseudarthrosis: Consensus from the children’s tumor foundation NF1 bone abnormalities consortium. J Pediatr Orthop 2013;33:269-75. [Google Scholar | PubMed]
- 12.Heervä E, Koffert A, Jokinen E, Kuorilehto T, Peltonen S, Aro HT, et al. A controlled register-based study of 460 neurofibromatosis 1 patients: Increased fracture risk in children and adults over 41 years of age. J Bone Miner Res 2012;27:2333-7. [Google Scholar | PubMed]
- 13.Jalabert M, Ferkal S, Souberbielle JC, Sbidian E, Mageau A, Eymard F, et al. Bone status according to neurofibromatosis type 1 phenotype: A descriptive study of 60 women in France. Calcif Tissue Int 2021;108:738-45. [Google Scholar | PubMed]
- 14.Lammert M, Kappler M, Mautner VF, Lammert K, Störkel S, Friedman JM, et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int 2005;16:1161-6. [Google Scholar | PubMed]
- 15.Heervä E, Leinonen P, Kuorilehto T, Peltonen S, Pöyhönen M, Väänänen K, et al. Neurofibromatosis 1-related osteopenia often progresses to osteoporosis in 12 years. Calcif Tissue Int 2013;92:23-7. [Google Scholar | PubMed]
- 16.Seitz S, Schnabel C, Busse B, Schmidt HU, Beil FT, Friedrich RE, et al. High bone turnover and accumulation of osteoid in patients with neurofibromatosis 1. Osteoporos Int 2010;21:119-27. [Google Scholar | PubMed]
- 17.Poyrazoğlu HG, Baş VN, Arslan A, Bastug F, Canpolat M, Per H, et al. Bone mineral density and bone metabolic markers’ status in children with neurofibromatosis type 1. J Pediatr Endocrinol Metab 2017;30:175-80. [Google Scholar | PubMed]
- 18.Schindeler A, Little DG. Recent insights into bone development, homeostasis, and repair in type 1 neurofibromatosis (Nf1). Bone 2008;42:616-22. [Google Scholar | PubMed]
- 19.Van der Velde GM, Hsu WS. Posterior tibial stress fracture: A report of three cases. Manipulative Physiol Ther 1999;22:341-6. [Google Scholar | PubMed]
- 20.Ryan EA, Reiss E. Oncogenous osteomalacia. Review of the world literature of 42 cases and report of two new cases. Am J Med 1984;77:501-12. [Google Scholar | PubMed]
- 21.Harvey JN, Gray C, Belchetz PE. Oncogenous osteomalacia and malignancy. Clin Endocrinol (Oxf) 1992;37:379-82. [Google Scholar | PubMed]
- 22.Makhlouf Y, Boussaid S, Ajlani H, Jemmali S, Rekik S, Sehli H, et al. A rare case of hypophosphataemic osteomalacia in von recklinghausen neurofibromatosis. Eur J Case Rep Intern Med 2021;8:002618. [Google Scholar | PubMed]
- 23.Obo T, Koriyama N, Tokito A, Ogiso K, Nishio Y. Neurofibromatosis type 1 associated with hypophosphatemic osteomalacia due to hypersecretion of fibroblast growth factor 23: A case report. J Med Case Rep 2020;14:56. [Google Scholar | PubMed]
- 24.Nagaratnam S, Karupiah M, Mustafa N. Debilitating pain and fractures: A rare case of hypophosphatemic osteomalacia with concomitant Vitamin D deficiency in neurofibromatosis type 1. J ASEAN Fed Endocr Soc 2020;35:105-8. [Google Scholar | PubMed]
- 25.Sahoo SK, Kushwaha P, Bharti N, Khedgikar V, Trivedi R, Agrawal V, et al. Elevated FGF23 in a patient with hypophosphatemic osteomalacia associated with neurofibromatosis type 1. Bone 2019;129:115055. [Google Scholar | PubMed]
- 26.Gupta A, Dwivedi A, Patel P, Gupta S. Hypophosphatemic osteomalacia in von Recklinghausen neurofibromatosis: Case report and literature review. Indian J Radiol Imaging 2015;25:63-6. [Google Scholar | PubMed]
- 27.Chadha M, Singh AP, Singh AP. Hypophosphataemic osteomalacia in neurofibromatosis. Acta Orthop Belg 2009;75:847-50. [Google Scholar | PubMed]
- 28.Soveid M. Tumor associated osteomalacia in neurofibromatosis: Case report and literature review. Med J Islam Repub Iran 2003;16:227-30. [Google Scholar | PubMed]
- 29.Konishi K, Nakamura M, Yamakawa H, Suzuki H, Saruta T, Hanaoka H, et al. Hypophosphatemic osteomalacia in von Recklinghausen neurofibromatosis. Am J Med Sci 1991;301:322-8. [Google Scholar | PubMed]
- 30.Weinstein RS, Harris RL. Hypercalcemic hyperparathyroidism and hypophosphatemic osteomalacia complicating neurofibromatosis. Calcif Tissue Int 1990;46:361-6. [Google Scholar | PubMed]
- 31.Suda A. Hypophosphatemic osteomalacia in von recklinghausen’s neurofibromatosis. J Bone Miner Metab 1989;7:18-23. [Google Scholar | PubMed]
- 32.Hogan DB, Anderson C, MacKenzie RA, Crilly RG. Hypophosphatemic osteomalacia complicating von Recklinghausen’s neurofibromatosis: Increase in spinal density on treatment. Bone 1986;7:9-12. [Google Scholar | PubMed]
- 33.Wattiaux MJ, De Vernejoul MC, Bletry O, Ulmann A, Rondier J, Godeau P. Maladie de Recklinghausen avec hypophosphoremie et osteomalacie. Rev Med Interne 1985;6:495-502. [Google Scholar | PubMed]
- 34.Kamiya N, Yamaguchi R, Aruwajoye O, Kim AJ, Kuroyanagi G, Phipps M, et al. Targeted disruption of NF1 in osteocytes increases FGF23 and osteoid with osteomalacia-like bone phenotype. J Bone Miner Res 2017;32:1716-26. [Google Scholar | PubMed]
- 35.Dunn IF, Heese O, Black PM. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J Neurooncol 2000;50:121-37. [Google Scholar | PubMed]
- 36.Nieto VG, Díaz BH, Subias JE, Alacio MT, Rodríguez JD, Sevilla JE, et al. Unilateral renal agenesis. New arguments about the genetic relationship between kidney malformations and urolithiasis. An Pediatr (Barc) 2016;85:240-6. [Google Scholar | PubMed]
- 37.Schorsch F, Pohlmeyer-Esch G, Lasserre-Bigot D. Unilateral renal agenesis/dysplasia associated with contralateral end-stage kidney disease in two Wistar rats. Eur J Vet Pathol 2001;7:127-34. [Google Scholar | PubMed]
- 38.Gou M, Ma Z. Osteomalacia, renal Fanconi syndrome, and bone tumor. J Int Med Res 2018;46:3487-90. [Google Scholar | PubMed]
- 39.Morlock G, Savary JP, Sebert JL, Durroux R, Gardiol JC. Osteomalacie vitamino-résistante hypophosphatémique associée à une neurofibromatose. Rev Rhum Mal Osteoartic 1982;49:125-30. [Google Scholar | PubMed]
- 40.Liu S, Gupta A, Quarles LD. Emerging role of fibroblast growth factor 23 in a bone-kidney axis regulating systemic phosphate homeostasis and extracellular matrix mineralization. Curr Opin Nephrol Hypertens 2007;16:329-35. [Google Scholar | PubMed]






Related Articles in Journal of Orthopaedic Case Reports
 April 1, 2026 Efficacy and Safety of Empirically Used Levonadifloxacin (Intravenous and Oral) in the Treatment of Patients with Bone and Joint Infection: A Multicentric, Investigator-Initiated Study
April 1, 2026 Efficacy and Safety of Empirically Used Levonadifloxacin (Intravenous and Oral) in the Treatment of Patients with Bone and Joint Infection: A Multicentric, Investigator-Initiated Study May 1, 2026 Aneurysmal Bone Cyst of Lateral Cuneiform: Challenge in Diagnosis and Management
May 1, 2026 Aneurysmal Bone Cyst of Lateral Cuneiform: Challenge in Diagnosis and Management July 1, 2026 Efficacy of Intra-articular Growth Factor Concentrate in Knee Osteoarthritis: A Randomized Controlled Trial
July 1, 2026 Efficacy of Intra-articular Growth Factor Concentrate in Knee Osteoarthritis: A Randomized Controlled Trial January 10, 2016 Treatment of Non Unions of Subtrochanteric Fractures Using an Anatomical Proximal Femur Locked Compression Plate – A Prospective Study of 13 Patients
January 10, 2016 Treatment of Non Unions of Subtrochanteric Fractures Using an Anatomical Proximal Femur Locked Compression Plate – A Prospective Study of 13 Patients