
1. Pleomorphic sarcoma may exhibit multiple divergent differentiation patterns, complicating diagnosis 2. Imaging may misleadingly suggest hematologic malignancy or metastasis 3. Broad immunohistochemical profiling is essential in atypical spindle-cell tumors 4. Multidisciplinary management significantly improves diagnostic accuracy and planning.
Dr. Arnav P Rathod, Department of Orthopedics, Dr. Panjabrao Deshmukh Memorial Medical College and Hospital, Amravati, Maharashtra, India. E-mail: rarnav95@yahoo.in
Abstract
Introduction: Pleomorphic sarcoma (PS) is a high-grade soft-tissue malignancy with marked morphologic variability. Divergent differentiation is recognized, but dual smooth-muscle and osteogenic lineage differentiation is exceedingly rare and may mimic other malignancies.
Case Report: A 52-year-old male presented with progressive low-back pain, weight loss, and an enlarging iliac mass. Magnetic resonance imaging showed multiple lytic lesions suggestive of plasma-cell dyscrasia; however, serum electrophoresis was negative. Histopathology revealed high-grade PS, and immunohistochemistry demonstrated simultaneous positivity for smooth-muscle actin and osteocalcin, confirming dual smooth-muscle and osteosarcomatous differentiation. The patient was referred to oncology for multidisciplinary management.
Conclusion: PS with dual divergent differentiation represents a rare diagnostic challenge. Clinical and imaging features may falsely suggest hematologic malignancy, underscoring the necessity of correlating radiology, histopathology, and immunohistochemistry.
Keywords: Pleomorphic sarcoma, divergent differentiation, smooth-muscle actin, osteocalcin, iliac lesion, diagnostic dilemma.
Pleomorphic sarcoma (PS), formerly classified as malignant fibrous histiocytoma when described by O’Brien and Stout in 1964 [1], is a high-grade mesenchymal malignancy known for extreme morphologic pleomorphism. Although reclassified under “Undifferentiated Pleomorphic Sarcoma (UPS)” in the present World Health Organization soft tissue and bone tumor classification [2], it remains one of the most commonly encountered adult soft-tissue sarcomas.
One reason for diagnostic difficulty is the occasional presence of divergent differentiation, wherein tumor cells display smooth-muscle, cartilaginous, or osteoid features [3]. Most tumors show only one such lineage; dual differentiation – such as simultaneous smooth-muscle and osteogenic elements – is extremely rare and can closely mimic leiomyosarcoma, osteosarcoma, or metastasis [4,5].
We report a rare case of PS demonstrating both smooth-muscle and osteosarcomatous differentiation, with initial imaging strongly suggesting plasma-cell dyscrasia, illustrating the need for broad diagnostic consideration.
A 52-year-old male presented with 6 months of worsening low-back pain, generalized weakness, and weight loss. A gradually enlarging swelling over the left iliac region had developed in the preceding weeks. He denied prior trauma, chronic illness, or substance use.
On examination, a firm, tender, irregular mass was palpated over the left iliac fossa and lumbar region. Straight leg raise reproduced radicular pain, but motor and sensory functions were intact. Distal neurovascular examination was normal (Fig. 1).
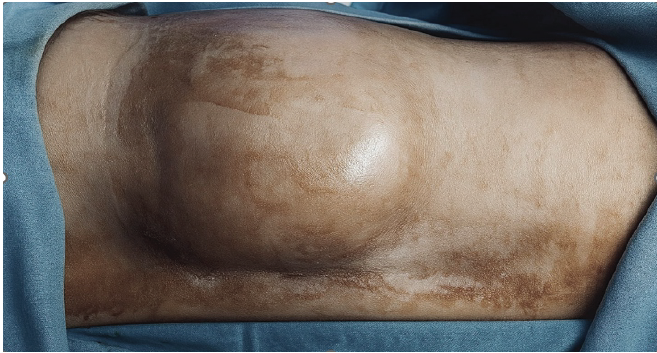
Figure 1:Clinical photograph showing a large, well-defined swelling over the left iliac and lumbar
region.
Investigations
Imaging
Magnetic resonance imaging (MRI) of the pelvis and spine revealed multiple lytic destructive lesions involving the right iliac bone and vertebral bodies, raising suspicion of plasma-cell dyscrasia or metastatic malignancy. The lesions demonstrated heterogeneous signal intensity with cortical destruction and soft-tissue extension. Post-contrast sequences showed heterogeneous enhancement, and areas of diffusion restriction suggested high cellularity.
Laboratory findings
- Serum protein electrophoresis: Negative for M-protein
- Serum calcium: Elevated.
Given conflicting imaging and laboratory findings, a biopsy was pursued.
Histopathology
Biopsy showed:
- Sheets and fascicles of highly pleomorphic spindle cells
- Hyperchromatic to vesicular nuclei with prominent nucleoli
- Brisk mitoses
- Areas of necrosis and hemorrhage.
These findings were consistent with a high-grade PS.
Imprint cytology supported a malignant spindle-cell process (Fig. 2).
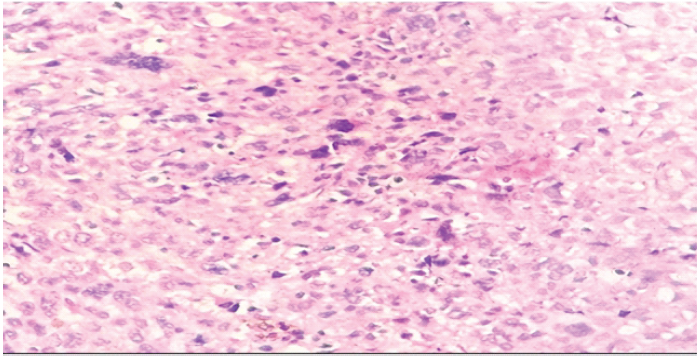
Figure 2: Hematoxylin and eosin section demonstrating spindle cells with bizarre, pleomorphic nuclei
A qualitative predominance of pleomorphic spindle-cell component was noted, with focal areas demonstrating smooth-muscle and osteogenic differentiation; however, precise quantitative assessment was not feasible on core biopsy areas of malignant osteoid deposition were identified, characterized by eosinophilic extracellular matrix intimately associated with atypical tumor cells. Numerous atypical mitotic figures were observed, along with significant areas of necrosis.
Immunohistochemistry
- Smooth muscle actin (SMA): Positive (Smooth-muscle differentiation)
- Osteocalcin: Positive (osteogenic differentiation)
- Vimentin: Positive (mesenchymal origin).
This combination established the diagnosis of PS with dual smooth-muscle and osteosarcomatous differentiation [4,6].
Management and follow-up
Following histopathological confirmation of PS with dual divergent differentiation, the patient was evaluated by a multidisciplinary tumor board consisting of orthopedic oncology, medical oncology, radiation oncology, and radiology specialists. Contrast-enhanced computed tomography (CT) of the chest, abdomen, and pelvis was performed for staging and demonstrated no visceral metastasis. A whole-body positron emission tomography-CT was advised to further delineate metabolic activity and detect occult disease, as recommended in contemporary sarcoma staging protocols [7,8].
Given the tumor’s involvement of the iliac wing with extension toward the paraspinal musculature and the pelvic ring, wide local excision was considered; however, the extent of disease rendered limb-salvage resection surgically challenging with high morbidity. Therefore, the tumor board opted for a non-surgical oncologic approach, consisting of neoadjuvant chemotherapy followed by external-beam radiotherapy, in accordance with established European Society for Medical Oncology (ESMO) and National Comprehensive Cancer Network (NCCN) guidelines for high-grade, unresectable soft-tissue sarcomas [7,8].
The patient was started on doxorubicin-based chemotherapy, the standard first-line regimen for advanced soft-tissue sarcoma aimed at reducing tumor burden and improving local control [7]. Radiotherapy planning targeted a dose of 60–66 Gy, consistent with guideline-directed practice for achieving adequate local tumor control in high-grade sarcomas [7,8].
At the 6-week follow-up, the patient reported partial improvement in pain and local discomfort, with stabilization of serum calcium levels following supportive therapy. A repeat MRI was planned after completion of the initial chemotherapy cycles to assess radiologic response and reevaluate the feasibility of surgical intervention should significant tumor regression occurs.
Due to financial limitations and declining functional capacity, the patient elected to continue treatment closer to home. Telephone follow-up at 3 months suggested partial symptom control, although interval imaging could not be obtained. Long-term oncologic outcome remains unknown.
PS with dual smooth-muscle and osteosarcomatous differentiation represents a rare diagnostic challenge. Clinical and imaging features may falsely suggest hematologic malignancy, underscoring the necessity of correlating radiology, histopathology, and immunohistochemistry. The lytic destructive lesions on imaging correlated with areas of tumor necrosis and aggressive spindle-cell proliferation on histopathology, explaining the radiologic resemblance to plasma-cell dyscrasia.
This report represents a single case, and therefore, conclusions regarding broader applicability are limited. However, given the rarity of dual-lineage differentiation in PS, such reports contribute valuable insights to existing literature.
Molecular genetic analysis, such as next-generation sequencing or MDM2 amplification testing, was not performed due to resource limitations. However, the diagnosis was established based on histomorphology and immunohistochemistry, consistent with standard diagnostic practice in many centers.
Although an extended immunohistochemical panel (Desmin, Myogenin, SATB2, MDM2, CDK4, S100, Cytokeratin, and CD34) was not performed, the combined positivity for SMA and osteocalcin, along with vimentin expression and characteristic morphology, was sufficient to establish divergent differentiation and exclude major mimics.
Long-term follow-up data, including progression-free and overall survival, could not be obtained, representing a limitation of this report.
As only a core biopsy specimen was available, complete tumor architecture and margin status could not be assessed.
Ki-67 proliferation index was not assessed, which could have provided additional prognostic information.
Core biopsy sampling may not fully represent tumor heterogeneity, and additional differentiation components cannot be entirely excluded.
Alternative treatment strategies, such as palliative resection, arterial embolization, or enrolment in targeted therapy trials may be considered in selected cases, particularly when surgical resection is not feasible.
Few cases of PS demonstrating divergent differentiation have been reported in the literature, most commonly showing either smooth-muscle or osteogenic lineage in isolation. Thway and Jones described cases with myogenic differentiation that closely resembled leiomyosarcoma, while Tamiolakis et al. reported osteoid-producing variants mimicking extraskeletal osteosarcoma. Similarly, Yamada et al. highlighted smooth-muscle differentiation in retroperitoneal tumors with overlapping histologic features.
In contrast, the present case demonstrates simultaneous dual differentiation, a phenomenon that is exceedingly rare. Furthermore, unlike previously reported cases, this tumor uniquely mimicked plasma-cell dyscrasia both clinically and radiologically, creating a significant diagnostic dilemma. This highlights the importance of maintaining a broad differential diagnosis in patients presenting with multifocal lytic lesions, particularly when laboratory findings are inconclusive (Table 1).
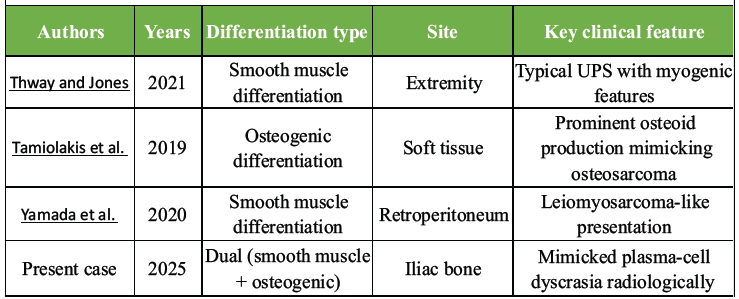
Table 1: Reported cases of pleomorphic sarcoma with divergent differentiation
Diagnostic pitfall: Mimicking plasma-cell dyscrasia
This patient’s presentation formed a classic diagnostic trap:
- Multiple lytic lesions → typical for multiple myeloma
- Hypercalcemia → frequently associated with myeloma
- Systemic symptoms → weight loss, fatigue
- MRI features similar to skeletal myeloma involvement [9].
However, the absence of monoclonal gammopathy, coupled with aggressive imaging features, mandated tissue diagnosis, highlighting the importance of not relying solely on radiology.
The lytic destructive lesions on imaging correlated with areas of tumor necrosis and aggressive spindle-cell proliferation on histopathology, explaining the radiologic resemblance to plasma-cell dyscrasia.
The radiologic appearance of multifocal lytic lesions with cortical destruction corresponded histologically to areas of aggressive spindle-cell proliferation, necrosis, and malignant osteoid deposition, further explaining the overlap with imaging features typically associated with plasma-cell dyscrasia.
Immunohistochemical consideration
The immunophenotype was key:
- SMA positivity → could mimic leiomyosarcoma
- Osteocalcin positivity → could mimic osteosarcoma
- Dual positivity → strongly suggests divergent PS rather than two separate tumors
- Vimentin → supports mesenchymal origin [6,10].
- The differential diagnosis was carefully evaluated based on immunohistochemical profile and clinicoradiological correlation, allowing exclusion of key mimickers such as myeloma, leiomyosarcoma, and osteosarcoma (Table 2).
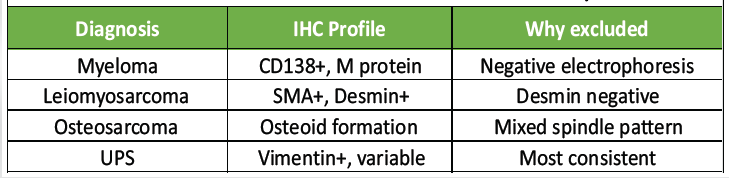
Table 2: Differential diagnosis of pleomorphic sarcoma with key
immunohistochemical features and reasons for exclusion in the present case.
Such tumors must be distinguished from:
- Leiomyosarcoma
- Conventional osteosarcoma
- Dedifferentiated liposarcoma
- Metastatic carcinoma
- UPS without divergence.
Clinical importance
PS is known for:
- High-grade and aggressive biological behavior
- High local recurrence rates
- Metastatic potential (often lungs) [7,8].
Management requires a multidisciplinary approach, including surgical oncology, medical oncology, and radiation oncology, per ESMO and NCCN guidelines [7,8].
PS with dual smooth-muscle and osteosarcomatous differentiation is an exceptionally rare variant that can closely mimic hematologic or metastatic disease. This case reinforces the need for careful clinicopathologic correlation and comprehensive immunohistochemical evaluation. Early recognition and prompt referral to a sarcoma specialty team are essential.
Pleomorphic sarcoma with dual divergent differentiation can closely mimic hematologic malignancy on imaging. When clinical, biochemical, and radiologic features are discordant, biopsy with comprehensive immunohistochemical evaluation is essential to avoid misdiagnosis.
References
- 1. O’Brien JE, Stout AP. Malignant fibrous xanthomas. Cancer 1964;17:1445-55. [Google Scholar] [PubMed]
- 2. WHO. WHO classification of tumours editorial board. In: Soft Tissue and Bone Tumours. 5th ed. Lyon: IARC; 2020. [Google Scholar] [PubMed]
- 3. Miettinen M. Modern soft-tissue pathology: Classification, markers, and prognosis. Mod Pathol 2014;27 Suppl 1:S17-29. [Google Scholar] [PubMed]
- 4. Thway K, Jones RL. Undifferentiated pleomorphic sarcoma: Diagnostic updates and advances. Histopathology 2021;79:348-61. [Google Scholar] [PubMed]
- 5. Tamiolakis D, Venizelos I, Nikolaidou S, Lambropoulou M, Papadopoulos N, Koutsougeras G, et al. Pleomorphic sarcoma with osteoid differentiation: Diagnostic pitfalls. Indian J Pathol Microbiol 2019;62:123-6. [Google Scholar] [PubMed]
- 6. Kyle RA, Gertz MA, Witzig TE, Lust JA, Lacy MQ, Dispenzieri A, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003;78:21-33. [Google Scholar] [PubMed]
- 7. Hornick JL. Practical immunohistochemistry in soft-tissue tumors. Arch Pathol Lab Med 2014;138:471-90. [Google Scholar] [PubMed]
- 8. Yamada Y, Yamamoto H, Kohashi K, Oda Y, Iwamoto Y, Taguchi T, et al. Clinicopathologic analysis of pleomorphic sarcoma with smooth muscle differentiation. Virchows Arch 2020;476:545-52. [Google Scholar] [PubMed]
- 9. Casali PG, Miah AB, Dei Tos AP, Abecassis N, Bajpai J, Bauer S, et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up soft tissue and visceral sarcomas: ESMO-EURACAN guidelines. Ann Oncol 2021;32:1348-65. [Google Scholar] [PubMed]
- 10. NCCN. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Soft Tissue Sarcoma. Ver. 1. United States: NCCN; 2024. [Google Scholar] [PubMed]





Related Articles in Journal of Orthopaedic Case Reports
 July 1, 2026 Diagnostic Dilemma in a Distal Femoral Intramedullary Lesion: Enchondroma Mimicking Low-Grade Chondrosarcoma with Discordant Biopsy Findings: A Case Report
July 1, 2026 Diagnostic Dilemma in a Distal Femoral Intramedullary Lesion: Enchondroma Mimicking Low-Grade Chondrosarcoma with Discordant Biopsy Findings: A Case Report June 1, 2026 Cortical Lytic Lesion of the Proximal Tibia in an 8-Year-Old Child: A Pediatric Diagnostic Conundrum
June 1, 2026 Cortical Lytic Lesion of the Proximal Tibia in an 8-Year-Old Child: A Pediatric Diagnostic Conundrum June 1, 2026 Atypical Bilateral Cystic Foot Swellings with Osteomyelitis-Like Features: A Diagnostic Dilemma
June 1, 2026 Atypical Bilateral Cystic Foot Swellings with Osteomyelitis-Like Features: A Diagnostic Dilemma February 1, 2026 Resolving the Diagnostic Dilemma: Decoding the Pre-operative Radiographic Signs for Predicting Modified Gartland Type IV Supracondylar Fractures of the Humerus in Children
February 1, 2026 Resolving the Diagnostic Dilemma: Decoding the Pre-operative Radiographic Signs for Predicting Modified Gartland Type IV Supracondylar Fractures of the Humerus in Children