
To effectively diagnose and treat infantile fibrosarcoma of the bone among the pediatric population, it is crucial to maintain a high level of suspicion for accurate diagnosis and to adopt a comprehensive, multidisciplinary treatment plan.
Dr. Said Saghieh, Department of Orthopaedic Surgery, American University of Beirut Medical Center, Cairo Street, Hamra, Beirut 1107 2020, Lebanon. E-mail: ss15@aub.edu.lb
Abstract
Introduction: In adults, fibrosarcoma (FS) of the bone is a rare occurrence. Infantile FS, particularly in the distal radius, is an exceedingly uncommon tumor and, to the best of our knowledge, has not yet been documented in the literature. In the subsequent report, we present a case involving a 2-year-old male diagnosed with primary FS of the distal radius.
Case Report: We hereby report the case of a 2-year-old Caucasian boy presenting with primary bone FS in the distal radius. X-rays revealed an osseous mass with an extraosseous component. MRI showed heterogeneous enhancement, suggestive of non-liquefied necrosis or possible fibrosis within the extraosseous soft-tissue component. The patient underwent a resection of the tumor, followed by central translocation of the ulna.
Conclusion: Managing infantile FS of the bone requires a multidisciplinary approach. A high index of suspicion is crucial for diagnosing the tumor. Further studies are needed to enhance our approach and management of infantile FS of the bone.
Keywords: Fibrosarcoma, Infantile, Surgery.
In adults, fibrosarcoma (FS) of the bone is an infrequent entity, comprising 5% of primary bone tumors. Infantile FS of the distal radius is exceptionally rare and, to the best of our knowledge, has not yet been reported in the literature. In this case report, we discuss a 2-year-old male patient presenting with primary FS of the distal radius. We will also review the literature and current management strategies, with an emphasis on the importance of adopting a multidisciplinary approach.
A 2-year-old male presented with a distal radius tumor on the left side that had been growing for 8 months. He presented with a pathological fracture, which was treated conservatively and healed 4 weeks after the injury. The mass had been noticed a couple of weeks before the fall but was not investigated until the patient came to the emergency department due to his fall. The family history was negative for consanguineous parents; however, it revealed that the mother had pre-eclampsia during her pregnancy. Upon physical examination, he had an obviously palpable, tender lesion with no erythema. He had no fever, no lymphadenopathy, and no upper respiratory symptoms. Basic laboratory investigations were within normal ranges, and the lactate dehydrogenase level was normal. Both oral and written informed consents were obtained from the legal guardians regarding the case study and future publications. X-ray imaging showed the osseous origin of the mass with an extraosseous component (Fig. 1a and b).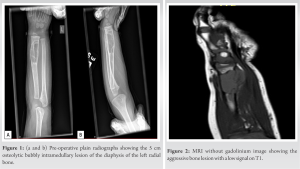 It revealed a 5 cm craniocaudally expansile, osteolytic, bubbly intramedullary lesion of the diaphysis of the left radial bone, extending to the metaphysis. The lesion displayed slow-growing features such as septations and a non-destructive cortical invasion. There were some cortical erosions and disruption, but no significant periosteal reaction. Proximally, there was a wide zone of transition; however, distally, only a narrow transition zone was present. It was difficult to determine whether the lesion was benign or malignant. The opacity between the radius and the ulna suggested extraosseous infiltration, which prompted consideration of a malignant etiology. An MRI was consequently ordered to further investigate the soft-tissue component. MRI images showed an aggressive, destructive bone lesion with a low signal on T1 (Fig. 2) and a high signal on STIR (Fig. 3a and b), involving the mid and distal radius with an associated soft-tissue component measuring 5.3 × 2.3 × 2.1 cm. The images showed restricted diffusion with an associated adjacent periosteal reaction.
It revealed a 5 cm craniocaudally expansile, osteolytic, bubbly intramedullary lesion of the diaphysis of the left radial bone, extending to the metaphysis. The lesion displayed slow-growing features such as septations and a non-destructive cortical invasion. There were some cortical erosions and disruption, but no significant periosteal reaction. Proximally, there was a wide zone of transition; however, distally, only a narrow transition zone was present. It was difficult to determine whether the lesion was benign or malignant. The opacity between the radius and the ulna suggested extraosseous infiltration, which prompted consideration of a malignant etiology. An MRI was consequently ordered to further investigate the soft-tissue component. MRI images showed an aggressive, destructive bone lesion with a low signal on T1 (Fig. 2) and a high signal on STIR (Fig. 3a and b), involving the mid and distal radius with an associated soft-tissue component measuring 5.3 × 2.3 × 2.1 cm. The images showed restricted diffusion with an associated adjacent periosteal reaction.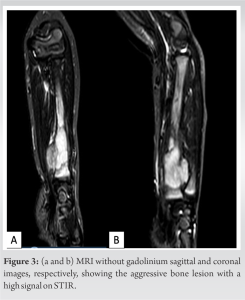 A dense enhancement within the bone lesion, along with heterogeneous enhancement, suggested non-liquefied necrosis or possible fibrosis within the extraosseous soft-tissue component (Fig. 4).
A dense enhancement within the bone lesion, along with heterogeneous enhancement, suggested non-liquefied necrosis or possible fibrosis within the extraosseous soft-tissue component (Fig. 4).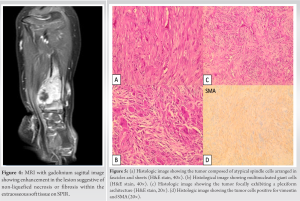 Both a CT and a PET scan of the chest were negative for metastasis. The differential diagnosis, based on clinical and radiological parameters, included Ewing sarcoma, rhabdomyosarcoma, Langerhans cell histiocytosis, infantile myofibromatosis, and infantile FS. A CT-guided biopsy was performed. Histologically, the sections revealed a proliferation of spindle cells with elongated nuclei arranged in fascicles, separated by numerous blood vessels (Fig. 5a). Mild nuclear atypia was present, and few mitoses (<5 HPF) were observed; rare giant cells were also present (Fig. 5b). The immunohistochemical study showed proliferating cells positive for vimentin and SMA (Fig. 5d). CD95, CD34, S100, and BCL2 were negative. Ki67 labeled 10–15% of the nuclei. The prominent plexiform architecture and myofibroblastic and histiocytic immunophenotypes (positive for histiocytic markers and smooth muscle actin) introduced plexiform fibrohistiocytic tumor into the differential diagnosis (Fig. 5c). However, this differential is atypical given the tumor size, location, aggressive features, and the patient’s young age. Considering this atypical presentation, a discussion with musculoskeletal oncology experts was initiated, and the biopsy specimen was sent for additional molecular studies. Following next-generation sequencing results—which enable sequencing of the entire genome within a day [1], offering enhanced sequencing output for a concrete insight into the biological context of the disease mechanism [2]—a low-grade infantile fibromyxoid sarcoma of the bone emerged as the most likely diagnosis. Considering the location and invasion of the soft-tissue extension, along with the encroachment of the neurovascular bundle, the patient underwent surgical resection in the operating room. Radical resection, referring to the extensive surgical removal of a bone tumor—in this case, infantile FS—aimed to eliminate the entire tumor and some surrounding healthy tissue, referred to as the “margin,” to ensure no malignant cells remained. This involved performing two osteotomies or surgical cuts into the bone. The first osteotomy was executed 5 cm proximal to the growth plate, with an additional centimeter allowed to ensure free margins, thus minimizing the risk of leaving cancerous cells near the growth plate—a critical area for growing bone in pediatric patients. The second osteotomy, distal to the first and 1 cm proximal to the growth plate, served to bracket the tumor and its associated tissues for complete removal (Fig. 6a and b).
Both a CT and a PET scan of the chest were negative for metastasis. The differential diagnosis, based on clinical and radiological parameters, included Ewing sarcoma, rhabdomyosarcoma, Langerhans cell histiocytosis, infantile myofibromatosis, and infantile FS. A CT-guided biopsy was performed. Histologically, the sections revealed a proliferation of spindle cells with elongated nuclei arranged in fascicles, separated by numerous blood vessels (Fig. 5a). Mild nuclear atypia was present, and few mitoses (<5 HPF) were observed; rare giant cells were also present (Fig. 5b). The immunohistochemical study showed proliferating cells positive for vimentin and SMA (Fig. 5d). CD95, CD34, S100, and BCL2 were negative. Ki67 labeled 10–15% of the nuclei. The prominent plexiform architecture and myofibroblastic and histiocytic immunophenotypes (positive for histiocytic markers and smooth muscle actin) introduced plexiform fibrohistiocytic tumor into the differential diagnosis (Fig. 5c). However, this differential is atypical given the tumor size, location, aggressive features, and the patient’s young age. Considering this atypical presentation, a discussion with musculoskeletal oncology experts was initiated, and the biopsy specimen was sent for additional molecular studies. Following next-generation sequencing results—which enable sequencing of the entire genome within a day [1], offering enhanced sequencing output for a concrete insight into the biological context of the disease mechanism [2]—a low-grade infantile fibromyxoid sarcoma of the bone emerged as the most likely diagnosis. Considering the location and invasion of the soft-tissue extension, along with the encroachment of the neurovascular bundle, the patient underwent surgical resection in the operating room. Radical resection, referring to the extensive surgical removal of a bone tumor—in this case, infantile FS—aimed to eliminate the entire tumor and some surrounding healthy tissue, referred to as the “margin,” to ensure no malignant cells remained. This involved performing two osteotomies or surgical cuts into the bone. The first osteotomy was executed 5 cm proximal to the growth plate, with an additional centimeter allowed to ensure free margins, thus minimizing the risk of leaving cancerous cells near the growth plate—a critical area for growing bone in pediatric patients. The second osteotomy, distal to the first and 1 cm proximal to the growth plate, served to bracket the tumor and its associated tissues for complete removal (Fig. 6a and b).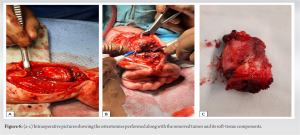 Following the osteotomies, the resection concluded with the total removal of the tumor and its soft-tissue components, including muscle, fat, and connective tissues (Fig. 6c). To further stabilize the surgical site and preserve the function of the radiocarpal joint, central translocation of the ulna was performed, with fixation achieved using K-wires, followed by the application of a long arm cast (Fig. 7a and b).
Following the osteotomies, the resection concluded with the total removal of the tumor and its soft-tissue components, including muscle, fat, and connective tissues (Fig. 6c). To further stabilize the surgical site and preserve the function of the radiocarpal joint, central translocation of the ulna was performed, with fixation achieved using K-wires, followed by the application of a long arm cast (Fig. 7a and b).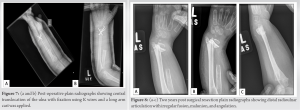 This comprehensive, multi-step surgical approach aimed to eradicate all potential sources of cancerous cells, stabilize the affected area, minimize the risk of recurrence, and maximize the chances of a complete cure. The patient’s post-operative course initially unfolded smoothly; however, the presence of positive surgical margins necessitated additional treatment. Consequently, the patient underwent adjuvant chemotherapy and radiotherapy. Four months post-resection surgery, the K-wires were removed due to suspected osteomyelitis. An 8-week treatment regimen of meropenem and teicoplanin was administered to address the infection. The oncological treatment encompassed a total of six chemotherapy cycles and 30 radiation therapy sessions. Specifically, the chemotherapy regimen included ifosfamide (Holoxan) at a dose of 3000 mg/m^2 IV every 24 hours and doxorubicin (Adriamycin) at a dose of 30 mg/m^2 IV every 24 hours, administered daily with the exception of weekends. Two years post-radical resection and subsequent treatments, a deformity developed in the patient’s wrist. Imaging revealed irregular fusion across the distal radioulnar articulation between the distal ulna and the remaining distal radial metaphysis and epiphysis. Malunion and angulation were also observed (Fig. 8a-c). These issues were successfully addressed through an osteotomy of the distal radius, followed by the application of an Ilizarov fixator. Post-Ilizarov fixation imaging indicated improved alignment, although evaluation was constrained due to the overlying metallic hardware (Fig. 9).
This comprehensive, multi-step surgical approach aimed to eradicate all potential sources of cancerous cells, stabilize the affected area, minimize the risk of recurrence, and maximize the chances of a complete cure. The patient’s post-operative course initially unfolded smoothly; however, the presence of positive surgical margins necessitated additional treatment. Consequently, the patient underwent adjuvant chemotherapy and radiotherapy. Four months post-resection surgery, the K-wires were removed due to suspected osteomyelitis. An 8-week treatment regimen of meropenem and teicoplanin was administered to address the infection. The oncological treatment encompassed a total of six chemotherapy cycles and 30 radiation therapy sessions. Specifically, the chemotherapy regimen included ifosfamide (Holoxan) at a dose of 3000 mg/m^2 IV every 24 hours and doxorubicin (Adriamycin) at a dose of 30 mg/m^2 IV every 24 hours, administered daily with the exception of weekends. Two years post-radical resection and subsequent treatments, a deformity developed in the patient’s wrist. Imaging revealed irregular fusion across the distal radioulnar articulation between the distal ulna and the remaining distal radial metaphysis and epiphysis. Malunion and angulation were also observed (Fig. 8a-c). These issues were successfully addressed through an osteotomy of the distal radius, followed by the application of an Ilizarov fixator. Post-Ilizarov fixation imaging indicated improved alignment, although evaluation was constrained due to the overlying metallic hardware (Fig. 9). The patient is currently faring well and has been discharged from the hospital in stable condition.
The patient is currently faring well and has been discharged from the hospital in stable condition.
Primary fibrosarcoma (FS) is an infrequent entity, comprising less than 5% of all primary bone tumors. Adult-type bone FS, a rare and malignant tumor originating from fibroblasts, predominantly affects adults and, only on very rare occasions, children, exhibiting an inconsistent response to therapy [3,4,5]. Infantile fibrosarcoma (IFS) most commonly involves the distal extremities, followed by the head, neck, and trunk, while the adult type is more frequently found in the thigh [6,7,8]. IFS, a rare malignant mesenchymal neoplasm derived from fibroblasts, has an incidence rate of fewer than 0.2 cases per million in the pediatric age group [9,10]. It primarily affects children within the first 2 years of life; however, it can be present at birth and can also develop during early childhood [9,11]. The most commonly recommended treatment is surgical resection [7]. To the best of our knowledge, infantile primary FS of the bone has not yet been reported in the literature, and its management has not been thoroughly investigated. Maintaining a high index of suspicion is crucial when faced with an unorthodox presentation of an infantile bony tumor. Previous studies have reported several FS cases of the bone with various suggested management algorithms. However, the aforementioned reported cases did not include young children. Although instances of regression following chemotherapy—without the need for any surgical intervention—have been reported, surgical resection remains the most commonly recommended treatment [12]. To date, all reported cases of primary FS in bone have involved either adults or children above 7 years of age. In their 2021 paper, Kosemehmetoglu et al. reviewed all reported cases of primary FS of the bone, identifying a total of 9 cases. The patients had a mean age of 39 years, with ages ranging from 14 to 71 years, and consisted of 5 males and 4 females [13]. Additionally, a retrospective study by Papagelopoulos et al. investigated 92 cases of primary FS of the bone and discussed their outcomes. Patients who underwent adequate radical resection demonstrated a better prognosis than those treated with intralesional or marginal resection. The study emphasized that further investigations are needed to explore the role of perioperative chemotherapy in patients with FS, concluding that age >40, tumors in the axial skeleton, and high-grade tumors are pivotal risk factors that negatively impact overall survival [5]. However, none of the patients in the series was under 5 years old.
A multidisciplinary approach is essential for managing infantile FS of the bone, and maintaining a high index of suspicion is crucial for diagnosing this tumor in the pediatric population. Additional studies are warranted to enhance the approach and management of infantile FS of the bone.
Developing a comprehensive strategy that addresses multiple aspects is a must for an effective infantile FS of bone treatment. Furthermore, keeping a vigilant approach is necessary to accurately identify this tumor in children. Further studies are required to enhance our knowledge and improve the management of infantile FS of the bone.
References
- 1.Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Child Educ Pract Ed 2013;98:236-8. [Google Scholar | PubMed]
- 2.Kumar KR, Cowley MJ, Davis RL. Next-generation sequencing and emerging technologies. Semin Thromb Hemost 2019;45:661-73. [Google Scholar | PubMed]
- 3.Bridge J, Hogendoorn PC, Mertens F. WHO Classification of Tumours of Soft Tissue and Bone. France: International Agency for Research on Cancer; 2013. [Google Scholar | PubMed]
- 4.Kilaikode S, Kuril S, Sedrak A, Sadanandan S. Sclerosing epithelioid fibrosarcoma of the parietal bone and adjacent meninges in an adolescent: A case report. World J Oncol 2013;4:255-7. [Google Scholar | PubMed]
- 5.Papagelopoulos PJ, Galanis E, Frassica FJ, Sim FH, Larson DR, Wold LE. Primary fibrosarcoma of bone. Outcome after primary surgical treatment. Clin Orthop Relat Res 2000;373:88-103. [Google Scholar | PubMed]
- 6.Chung EB, Enzinger FM. Infantile fibrosarcoma. Cancer 1976;38:729-39. [Google Scholar | PubMed]
- 7.Himori K, Hatori M, Watanabe M, Moriya T, Ogose A, Hashimoto H, et al. Infantile fibrosarcoma of thigh--a case report. Ups J Med Sci 2005;110:85-93. [Google Scholar | PubMed]
- 8.Soule EH, Pritchard DJ. Fibrosarcoma in infants and children: A review of 110 cases. Cancer 1977;40:1711-21. [Google Scholar | PubMed]
- 9.Priya M, Singh P, Malhotra M, Angral S, Varshney S, Bhardwaj A, et al. Cervical infantile fibrosarcoma: A rare cause of paediatric parapharyngeal neck mass. Autops Case Rep 2020;10:e2020189. [Google Scholar | PubMed]
- 10.Thebaud E, Mezel A, Leroy X, Orbach D. Fibrosarcoma in children and adolescents: Different entities for the same name. Bull Cancer 2012;99:715-22. [Google Scholar | PubMed]
- 11.Coffin CM, Jaszcz W, O’Shea PA, Dehner LP. So-called congenital-infantile fibrosarcoma: Does it exist and what is it? Pediatr Pathol 1994;14:133-50. [Google Scholar | PubMed]
- 12.Sait SF, Danzer E, Ramirez D, LaQuaglia MP, Paul M. Spontaneous regression in a patient with infantile fibrosarcoma. J Pediatr Hematol Oncol 2018;40:e253-5. [Google Scholar | PubMed]
- 13.Kosemehmetoglu, K., Ardic F, Kilpatrick SE, Aydingoz U, Sumathi VP, Michal M. Sclerosing epithelioid fibrosarcoma of bone: morphological, immunophenotypical, and molecular findings of 9 cases. Virchows Arch, 2021. 478(4):767-777. [Google Scholar | PubMed]




Related Articles in Journal of Orthopaedic Case Reports
 October 10, 2023 Common Peroneal Nerve Palsy Due to Chronic Exertional Compartment Syndrome after Military Training: A Case Report
October 10, 2023 Common Peroneal Nerve Palsy Due to Chronic Exertional Compartment Syndrome after Military Training: A Case Report September 10, 2023 Iliofemoral Arthrodesis as a Reconstructive Surgical Technique in the Management of Malignant Periacetabular Tumors: Case Series
September 10, 2023 Iliofemoral Arthrodesis as a Reconstructive Surgical Technique in the Management of Malignant Periacetabular Tumors: Case Series August 6, 2024 Uncommon Expression of Osteochondroma in the Wrist: A Case Report
August 6, 2024 Uncommon Expression of Osteochondroma in the Wrist: A Case Report July 10, 2020 Subcapital Fracture with Avascular Necrosis of the Whole Femoral Head After Fixation of an Intertrochanteric Fracture: A Case Report
July 10, 2020 Subcapital Fracture with Avascular Necrosis of the Whole Femoral Head After Fixation of an Intertrochanteric Fracture: A Case Report