
Ewing’s sarcoma requires a global, multidisciplinary approach with the primary objective of patient survival and autonomy in the growing child.
Dr. Mohamed Khiareddine, Department of Orthopaedic Surgery, Sahloul Hospital, 4054 Sousse, Tunisia. E-mail: mohamedkhiareddine99@gmail.com
Abstract
Introduction: Ewing’s sarcoma is the second most common primary malignant bone tumor in children and adolescents, after osteosarcoma. It is a rare tumor, with the axial skeleton being the preferred site of development, followed by the long bones. Diagnosis is evoked by imaging and confirmed by histology. Treatment is based on intensive chemotherapy with local surgical treatment in operable forms, and in some cases, radiotherapy. There are only a few sporadic cases in the literature describing distal fibular localization.
Case Report: We report the case of a 7-year-old child who presented with pain on the lateral aspect of the left ankle without local inflammatory signs. Radiological findings revealed a metaphyseal-diaphyseal osteolytic tissue process of the left fibula, and histology concluded that it was Ewing’s sarcoma. We proceeded with neoadjuvant polychemotherapy followed by segmental resection of the distal fibula with an anteroexternal tibial rod, plus tibiotalar, and talocalcaneal arthrodesis without recourse to radiotherapy.
Conclusion: The management of Ewing’s sarcoma is constantly evolving. Its distal fibular location in a growing limb makes it even more difficult. It must be personalized, multidisciplinary, and carried out in specialized centers.
Keywords: Ewing’s sarcoma, distal fibula, chemotherapy arthrodesis.
Ewing’s sarcoma was first described in 1921 [1]. It is the second most common primary malignant bone tumor in children and adolescents, after osteosarcoma [2]. Preferentially affecting the pelvis, spine, and rib cage, distal fibular localization is rare. Cross-sectional imaging plays an important role in diagnosis, following pathological examination [3]. Multidrug chemotherapy with surgery is often indicated in resectable forms, sometimes in combination with radiotherapy. We report the case of a 7-year-old child presenting with pain on the lateral aspect of the left ankle, in whom the radiological work-up revealed a metaphyseal-diaphyseal osteolytic tissue process of the left fibula, and histology concluded that it was Ewing’s sarcoma.
This is a 7-year-old child with no particular pathological history who consulted us in 2021 for left ankle pain with lameness, which had been evolving for 3 months in the context of apyrexia. A standard radiological workup showed an osteolytic image of the distal metaphysis of the left fibula with a Codman spur-type periosteal reaction (Fig. 1). An magnetic resonance imaging (MRI) scan showed a rapidly progressive aggressive process in the distal meta-physiodiaphysis of the left fibula, sparing the conjugation cartilage and with a signal abnormality that enhanced after gadolinium injection, associated with edema (Fig. 2). 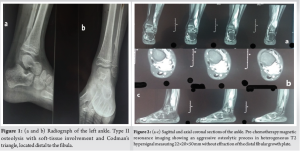
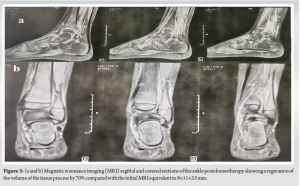
A bone scan revealed hyperfixation at the distal end of the fibula. A thoraco-abdomino-pelvic computed tomography (CT) scan returned without abnormality. An initial surgical biopsy via a posterolateral approach centered on the distal fibular metaphysis was performed, followed by a pathological examination which confirmed the malignant nature and concluded that the tumor was a highly proliferative round-cell tumor. Immunohistochemistry revealed cells diffusely expressing CD99, Fli-1, and NSE, while pancytokeratin and PS100 were negative, consistent with Ewing’s sarcoma. After a multidisciplinary staff meeting involving orthopedists, oncopediatricians, and radiotherapists, neoadjuvant chemotherapy was recommended in accordance with the EuroEWING 2012 protocol [4] with nine courses of vincristine, doxorubicin, cyclophosphamide, and ifosfamide etoposide. A post-chimico MRI was performed after the nineth treatment, showing a 70% reduction in tumor mass compared with the initial MRI of the distal fibular metaphyseal-diaphyseal osteolytic process, without extension to the tibia (Fig. 3).
Tumor resection was then programmed, based on a cookie-cutter excision of the tumor region with a healthy margin of 3 cm and a posterior hemidecortication of the tibia (Fig. 4).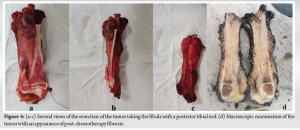
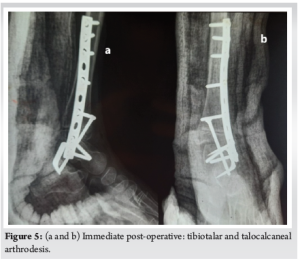
Tibiotalar arthrodesis with an AO plate followed by talocalcaneal arthrodesis with anteroposterior tibio-talo-calcaneal screw fixation reinforced by an external talocalcaneal staple was recommended (Fig. 5). The post-operative course was straightforward, with good skin healing, and bone consolidation was achieved after 2-month postoperatively. Weight-bearing was permitted at 2-month post-operative. Histological examination of the excision specimen showed post-chemotherapy fibrous remodeling without viable tumor cells (good responder) with surgical resection limits. We did not use radiotherapy.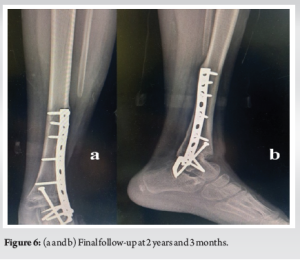
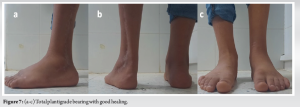
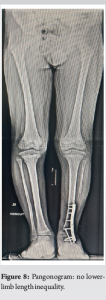
At present, at 2 years and 3 months post-therapy, there has been no local recurrence or secondary localization, no stigmata of local infection (Fig. 6), and a KITAOKA functional score [5] of 67/100 considered fair (given the arthrodesis) (Fig. 7), with no residual lower-limb length inequality (Fig. 8).
Ewing’s sarcoma is a rare, highly cancer, with most patients presenting with a priori micrometastases, since in the absence of systemic treatment, over 90% of patients die of disseminated disease [6,7]. The disease is most often diagnosed in the second decade of life. However, patients have presented as early as birth and as late as the eighth decade with tumors in almost every part of the body, in particular, the fibula, which constitutes a rare localization with a great surgical challenge in front of a growing limb with a percentage of 6.7% of all localizations [8,9]. The tumor results from a translocation between chromosomes 11 and 22, referred to as t (11; 22). The gene on chromosome 22 codes for the Ewing’s sarcoma protein, whose function is not well understood. The gene on chromosome 11, FLI1, activates and deactivates other genes [10]. Today, the primary diagnosis of bone pain, particularly in children, requires MRI, which has a high negative predictive value for malignant bone tumors [11]. However, standard X-rays are an essential first-line procedure, especially in view of Lodwic’s classification, which is still valid for osteolysis. CT scans are important for the assessment of extension, especially to the thorax, and bone scintigraphy for bone metastases. On MRI, Ewing’s sarcoma appears as a solid tumor mass in the bone, with T1 hyposignal and T2 hypersignal. There is often a sharp transition zone in the bony part of the tumor. The tumor shows peri-tumoral edema and gadolinium enhancement. PET scan is particularly useful for staging, restaging, and assessing therapeutic response in patients with Ewing’s tumor [12]. Pathological examination provides diagnostic confirmation, but the preferred biopsy method remains controversial, as no randomized controlled trial has yet been carried out to compare the surgical biopsy procedure with the percutaneous needle biopsy procedure [13]. Definitive diagnosis of the tumor should be made by biopsy in a sarcoma referral center, providing sufficient material for conventional histology, immunohistochemistry, and molecular pathology. Management must be multidisciplinary, personalized, and in specialized centers, based on neoadjuvant chemotherapy combined with surgery for operable local forms. Neoadjuvant chemotherapy is the mainstay of treatment for Ewing’s sarcoma [14]. The treatment plan generally consists of three stages: the first stage is initial cytoreduction with neoadjuvant chemotherapy to eradicate micro-metastases, facilitate local control by surgery with healthy margins, and prevent recurrence. The second stage is local treatment with surgery or radiotherapy. Finally, consolidation therapy is to eradicate occult residual disease and reduce the likelihood of tumor recurrence [15]. Surgery is the preferred complementary treatment in resectable forms. Only a few sporadic cases have been reported in the literature, where reconstruction using a contralateral fibular autograft after segmental resection, followed by arthrodesis, has been recommended [16]. For Norman–Taylor et al., fibulectomy alone for the treatment of distal fibular Ewing’s sarcoma has provided good results [17]. Although vascularized fibula autografting appears to be the most promising treatment, it seems to carry a high risk of fracture in a very active child [18]. We considered segmental resection followed by definitive tibiotalar and talocalcaneal arthrodesis to be an excellent alternative, enabling amputation to be avoided with less morbidity than other techniques. As for radiotherapy, the radiosensitivity of Ewing’s sarcoma has been recognized since it was first described by James Ewing [19]. Radiotherapy as an active therapeutic modality to ensure local control is used in inoperable forms or association with surgery, pre or postoperatively, and is also indicated in cases where surgery is incomplete, there is a poor response to chemotherapy or the tumor is difficult to access. Patients may also benefit from RT in a palliative setting [20].
Despite significant efforts in diagnostic and therapeutic strategies, many aspects of Ewing’s sarcoma remain elusive, underlining the ongoing need for internationally coordinated and joint translational research. At present, clinical decision-making is balanced between clinical facts and inter-individual concepts. Recent decades have shown that tumor-specific strategies are difficult to implement. Undirected chemotherapy still controls the disease in many patients but fails in far too many others. Prognosis depends essentially on two parameters: initial metastases and response to chemotherapy. That’s why multidisciplinary consultation meetings are always beneficial for both patient and clinician.
The management of Ewing’s sarcoma is burdensome for the patient and family, and although it involves doctors from different specialties, the outcome is not always certain. This tumor must remain a subject of study to facilitate management and prevent complications that can sometimes be harmful.
References
- 1.Euler E, Wilhelm K, Permanetter W, Kreusser T. Ewing’s sarcoma of the hand: Localization and treatment. J Hand Surg Am 1990;15:659-62. [Google Scholar | PubMed]
- 2.Anakwenze OA, Parker WL, Wold LE, Amrami KK, Amadio PC. Ewing’s sarcoma of the hand. J Hand Surg Eur Vol 2009;34:35-9. [Google Scholar | PubMed]
- 3.Javery O, Krajewski K, O’Regan K, Kis B, Giardino A, Jagannathan J, et al. A to Z of extraskeletal Ewing sarcoma family of tumors in adults: Imaging features of primary disease, metastatic patterns, and treatment responses. AJR Am J Roentgenol 2011;197:W1015-22. [Google Scholar | PubMed]
- 4.Anderton J, Moroz V, Marec-Bérard P, Gaspar N, Laurence V, Martín-Broto J, et al. International randomised controlled trial for the treatment of newly diagnosed EWING sarcoma family of tumours-EURO EWING 2012 protocol. Trials 2020;21:96. [Google Scholar | PubMed]
- 5.Kitaoka HB, Patzer GL. Analysis of clinical grading scales for the foot and ankle. Foot Ankle Int 1997;18:443-6. 6. Dahlin DC, Coventry MB, Scanlon PW. Ewing’s sarcoma. A critical analysis of 165 cases. J Bone Joint Surg Am 1961;43-A:185-92. [Google Scholar | PubMed]
- 6.Ewing J. The classic: Diffuse endothelioma of bone. Proceedings of the New York pathological society. 1921;12:17. Clin Orthop Relat Res 2006;450:25-7. [Google Scholar | PubMed]
- 7.Strimbu CV, Deacanu AB, Esanu AC, Luca AE, Cracana SN. Ewing sarcoma of fibula: A pediatric case of disease regression and bone regeneration. Case report and literature review. Curr Health Sci J 2020;46:207-12. [Google Scholar | PubMed]
- 8.Cotterill SJ, Ahrens S, Paulussen M, Jürgens HF, Voûte PA, Gadner H, et al. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol 2000;18:3108-14. [Google Scholar | PubMed]
- 9.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992;359:162-5. [Google Scholar | PubMed]
- 10.Daniel A Jr., Ullah E, Wahab S, Kumar V Jr. Relevance of MRI in prediction of malignancy of musculoskeletal system--a prospective evaluation. BMC Musculoskelet Disord 2009;10:125. [Google Scholar | PubMed]
- 11.Guimarães JB, Rigo L, Lewin F, Emerick A. The importance of PET/CT in the evaluation of patients with Ewing tumors. Radiol Bras 2015;48:175-80. [Google Scholar | PubMed]
- 12.Gerrand C, Bate J, Seddon B, Dirksen U, Randall RL, van de Sande M, et al. Seeking international consensus on approaches to primary tumour treatment in Ewing sarcoma. Clin Sarcoma Res 2020;10:21. [Google Scholar | PubMed]
- 13.Hustu HO, Holton C, James D Jr., Pinkel D. Treatment of Ewing’s sarcoma with concurrent radiotherapy and chemotherapy. J Pediatr 1968;73:249-51. [Google Scholar | PubMed]
- 14.Jain S, Kapoor G. Chemotherapy in Ewing’s sarcoma. Indian J Orthop 2010;44:369-77. [Google Scholar | PubMed]
- 15.Lenze U, Kasal S, Hefti F, Krieg AH. Non-vascularised fibula grafts for reconstruction of segmental and hemicortical bone defects following meta-/diaphyseal tumour resection at the extremities. BMC Musculoskelet Disord 2017;18:289. [Google Scholar | PubMed]
- 16.Norman-Taylor FH, Sweetnam DI, Fixsen JA. Distal fibulectomy for Ewing’s sarcoma. J Bone Joint Surg Br 1994;76:559-62. [Google Scholar | PubMed]
- 17.Mizoshiri N, Shirai T, Terauchi R, Tsuchida S, Mori Y, Katsuyama Y, et al. Limb saving surgery for Ewing’s sarcoma of the distal tibia: A case report. BMC Cancer 2018;18:503. [Google Scholar | PubMed]
- 18.Ewing J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA Cancer J Clin 1972;22:95-8. [Google Scholar | PubMed]
- 19.Rao AD, Chen Q, Ermoian RP, Alcorn SR, Figueiredo ML, Chen MJ, et al. Practice patterns of palliative radiation therapy in pediatric oncology patients in an international pediatric research consortium. Pediatr Blood Cancer 2017;64:e26589. [Google Scholar | PubMed]






Related Articles in Journal of Orthopaedic Case Reports
 February 1, 2025 Internal Plate Fixation of Comminuted Fracture of the Distal End of the Tibia and Fibula Using a Single-Incision Technique: A Novel Approach and a Series of Nine Cases
February 1, 2025 Internal Plate Fixation of Comminuted Fracture of the Distal End of the Tibia and Fibula Using a Single-Incision Technique: A Novel Approach and a Series of Nine Cases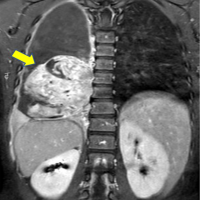 March 10, 2024 Philosophies and Surgical Techniques for Ewing’s Sarcoma of Spine with Review of Literature
March 10, 2024 Philosophies and Surgical Techniques for Ewing’s Sarcoma of Spine with Review of Literature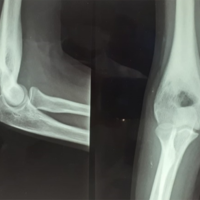 December 10, 2023 Tubercular Osteomyelitis Mimic Ewing’s Sarcoma
December 10, 2023 Tubercular Osteomyelitis Mimic Ewing’s Sarcoma June 10, 2023 Periosteal Osteosarcoma of the Distal Shaft of Fibula: Case Report on Rare Entity
June 10, 2023 Periosteal Osteosarcoma of the Distal Shaft of Fibula: Case Report on Rare Entity