
We report the case of a 19-year-old Caucasian male who developed progressive pseudorheumatoid dysplasia with all the symptoms described in the literature and underwent replacement of multiple joints at an early age for this disorder.
Dr. Antoine Outrequin, Department of Orthopedic and Trauma, Cliniques Universitaires Saint-Luc UCL, UCLouvain, Bruxelles, Belgium. E-mail: antoine_outrequin@hotmail.fr
Abstract
Introduction: Progressive pseudorheumatoid dysplasia is a rare autosomal recessive disorder caused by a mutation in the Wnt1-inducible signaling protein 3 gene, with few cases reported.
Case Report: We discuss the case of a 19-year-old Caucasian male patient with polyarticular involvement, including shoulders, elbows, wrists, hands, spine, hips, knees, and notably the ankles. Despite a well-conducted medical treatment, due to the rapid progression of his condition, the patient underwent bilateral total hip and knee arthroplasties, as well as left ankle replacement.
Conclusion: This case report highlights the importance of conducting the diagnosis of these rare diseases and the important place of joint replacements in the recovery of joint functions, even in young patients.
Keywords: Progressive pseudorheumatoid dysplasia, total arthroplasty, WISP3, mutation.
Progressive pseudorheumatoid dysplasia (PPRD) is a very rare autosomal recessive disorder caused by a mutation in the Wnt1-inducible signaling protein 3 (WISP3 gene) [1-3]. Symptoms usually begin between 3 and 8 years of age, but diagnosis often made between 10 and 20 years [4,5]. The child develops postural abnormalities, symmetric arthralgia, joint swelling, scoliosis, and difficulty walking. Inflammatory biological markers such as C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), rheumatoid factor (RF), anti-citrullinated, and anti-nuclear antibodies are normal in this disorder [1]. The main differential diagnosis is juvenile idiopathic arthritis (JIA) [1,6,7]. We report the case of a 19-year-old Caucasian male who developed PPRD with all the symptoms described in the literature and underwent replacement of multiple joints at an early age for this disorder.
The symptomatology began at around 3 to 4 years of age with difficulty to walk and radiographic evidence of delayed growth. At 7 years of age, he presented with muscular fatigue and painful bilateral coxalgia, limiting his walking perimeter to one km. Hyperlordosis, limping, five-degree flexion contracture of the knee, and flat feet were already present. At this age, he was already undergoing physiotherapy treatment several times a week. The patient was first diagnosed with spondyloperipheral dysplasia at the age of seven. PPRD was diagnosed at the age of 11 based on clinical and radiological evidence. A homozygous mutation of WISP3 was subsequently detected, confirming the diagnosis of DPPR. Ultrasounds of the shoulders, elbows, wrists, hips, knees, and ankles showed no synovial swelling or effusion at this stage. CRP, RF, anti-citrulline, and anti-nuclear antibodies consistently exhibit normal levels, thereby ruling out the possibility of a systemic inflammatory joint disorder in the diagnosis. At 12 years of age, a bone density scan revealed significant osteoporosis in the hips, and he walked with the aid of two crutches for short distances and used a manual wheelchair. At 17 years of age, the patient had stage four bilateral gonarthrosis, according to Kellgren and Laurence, with a fixed flessum, three degrees right varus, and two degrees left valgus (Fig. 1).
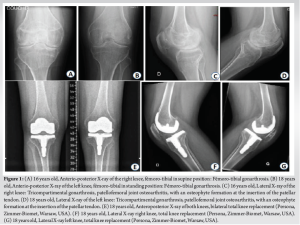
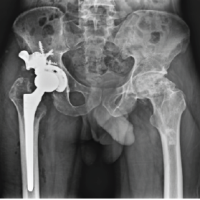
He underwent a right persona cemented total knee replacement (Zimmer-Biomet, Warsaw, USA), followed 13 months later by the same surgery on the left side. At the age of 18, the severity of bilateral coxarthrosis warrants a simultaneous bilateral total hip arthroplasty. This procedure involves the implantation of short-stem prostheses that utilize a ceramic on polyethylene bearings (Fig. 2).
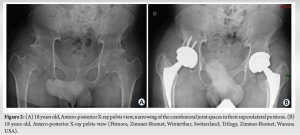
At this age, his adult height was 148.5 cm, and he weighed 59 kg. At 19 years of age, clinical evolution was characterized by spinal chondrodysplasia with disc protrusions leading to significant stenosis of the dural sac at Lumbar 1-Lumbar 2-Lumbar 3 (anteroposterior diameter of 5–6 mm) but without symptoms (Fig. 3).
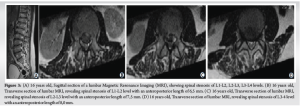
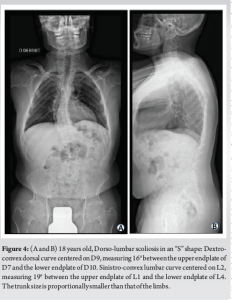
He also had dorsolumbar scoliosis with a left convexity of 25°, fusion of the Cervical 6-Cervical 7 posterior apophyses, and platyspondyly (Fig. 4).
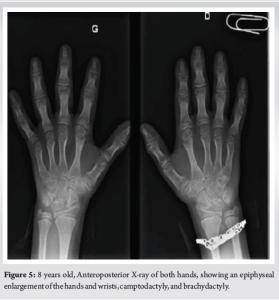
The cervical spine, shoulders, elbows, and wrists had limited mobility. We observed an epiphyseal enlargement of the hands and wrists, camptodactyly, and brachydactyly (Fig. 5).
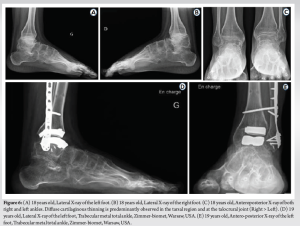
At the age of 20, severe bilateral osteoarthritis of the ankle joint is noted, more prominent on the left side, associated with epiphyseal enlargement of the base of the metatarsals bilaterally. The mobility of both ankle joints was reduced in all planes. Given the clinical and radiological situation, a surgical indication for bilateral total ankle replacement was proposed (Fig. 6). Currently, the patient is being treated with 1 g of calcium carbonate per day and D-cure at a dose of one to two ampoules per month, with the aim of maintaining bone mass and preventing the risk of fracture. The patient can walk without support for short distances. He reports persistent functional impairment related to non-replaced joints (foot, upper limb) and muscle weakness. He reports little relief from pain with the use of paracetamol and non-steroidal anti-inflammatory drugs (NSAIDs) and will most probably undergo other joint replacements in the upper limb in the future.
Our patient presents an advanced case of DPPR with involvement of all four limbs and the spine. After long-term physiotherapy management, surgical interventions were necessary as the final therapeutic option. This is an extremely rare disease, with few cases reported. The precise incidence is unknown but estimated to be 1/1,000,000 based on the incidence in the United Kingdom. The frequency is higher in Maghreb, Turkey, and India, as well as in the Middle East, and increases with consanguineous marriages [1-3, 8]. DPPR is part of the class ten osteochondrodysplasias, “other spondyloepi-(meta)physial dysplasia” family, according to the International Classification of Osteochondrodysplasias [4,9]. It is caused by a bi-allelic mutation of the WISP3 gene located on chromosome 6q22. It belongs to the CCN gene family, which encodes growth factors regulating cell proliferation, differentiation, and migration. The WISP3 gene is expressed in synoviocytes, chondrocytes, and mesenchymal cells. Its exact role is not yet understood. However, we know that it codes for a growth factor that promotes the production of type II collagen, aggrecan, and regulates chondrocyte proliferation and differentiation. Its absence does not interfere with skeletal morphology in utero, but it is essential for postnatal cartilage growth and homeostasis [1-3, 8]. The disease typically begins between three and 8 years of age with symmetrical joint stiffness, often at the hip level, as well as fatigue, abnormal posture, difficulty walking, swelling of the metacarpophalangeal joints, and occasionally pain. The stiffness then affects the metacarpophalangeal joints, hips, knees, and less commonly the cervical spine, shoulders, elbows, and wrists [1-3, 6, 10, 11]. Thoracic kyphosis and scoliosis develop during adolescence [1,11]. Growth is normal in early childhood but declines to a height below the third percentile [1,2]. The trunk size will be proportionally smaller than that of the limbs. Our patient presents all the aforementioned manifestations as well as ankle involvement. These arthropathies are characterized by the absence of synovitis with synovial swelling on ultrasound, magnetic resonance imaging (MRI), or biopsy [4,5]. Inflammatory markers such as CRP, ESR, and RF, as well as anti-citrullinated peptide and anti-nuclear antibodies, remain within normal values. In addition to clinical examination, lateral radiography of the spine helps to make the diagnosis [3,8,11]. Spondyloepiphyseal dysplasia, platyspondyly with irregular vertebral endplates throughout the spine, and thoracic kyphosis are observed. Radiography also shows a wide femoral neck with a squat head, broad tibial and femoral epiphyses, reduced joint space in the hips and knees, epiphyseal widening of the metacarpals and phalanges, and the presence of a megatrigonum bone in the foot radiograph, which is the result of excessive endochondral growth of the bone [8,10]. Bone scintigraphy and MRI can aid in the diagnostic approach [4,8]. Osteodensitometry will show very early osteopenia. The diagnosis can be confirmed by sequencing the WISP3 gene or by a skin biopsy [1,2]. The differential diagnosis is mainly made with JIA, which is more common with an incidence of 10–15/100,000. JIA responds very well to NSAIDs, methotrexate, and TNF inhibitors. Many misdiagnoses have led patients with PPRD to receive these treatments, which have shown no benefit in symptomatology or disease progression [1,2,6,7,11,12]. Note that Scheuermann’s disease, with an incidence ranging from 0.4% to 8% of the population, shares the same type of vertebral endplate abnormality located in the lower thoracic and lumbar regions, as well as thoracic kyphosis, making it the most common cause of kyphosis in adolescence. It should not be confused with lateral spine radiography [8, 10-13]. The treatment of DPPR remains palliative. First-line analgesics (paracetamol and NSAIDs) are quickly insufficient, as in our patient’s case. Some patients have chronic recourse to opioids to manage their pain. Physiotherapy will be intensive and aimed at maintaining joint mobility and improving posture through muscle strengthening and stretching [1,8]. Bisphosphonates have not shown any improvement in osteoporosis [1]. Hip and knee arthroplasties are generally required by the age of 30 [1,6,12]. A case of hip arthroplasty has been reported at the age of 17 [12]. The same goes for spinal surgery, which is at high risk of nerve and dural sac injuries due to adhesions and anatomical deformities [11]. Spinal surgery is not currently necessary for our patient despite significant deformities and spinal stenosis. Ankle involvement is rare in this pathology, and no cases of total ankle arthroplasty have been described to our knowledge nor have hip or knee arthroplasties at such a young age.
DPPR is a very rare autosomal recessive disease due to a mutation in the WISP3 gene. It is characterized by early joint involvement and bone growth disorders. No curative treatment has been described for this pathology, and treatment remains symptomatic to this day. Surgical interventions are frequent and early in the case of the patient discussed in this work. These patients should, therefore, be referred to a specialized environment from diagnosis, given the rarity of the pathology and the heavy management. A differential diagnosis of this pathology will be made with JIA and systemic pathologies involving joint and spinal involvement. Our patient was correctly diagnosed and adequately treated with early and intensive physiotherapy. Joint replacement was nevertheless required at an early stage to allow walking and weight bearing without significant pain.
Despite an early diagnosis and intensive physiotherapy, arthroplasty of the hips, knees, or ankles may become necessary before the second decade of life in this medical condition.
References
- 1.Torreggiani S, Torcoletti M, Campos-Xavier B, Baldo F, Agostoni C, Superti-Furga A, et al. Progressive pseudorheumatoid dysplasia: A rare childhood disease. Rheumatol Int 2018;39:441-52. [Google Scholar | PubMed]
- 2.Garcia Segarra N, Mittaz L, Campos-Xavier AB, Bartels CF, Tuysuz B, Alanay Y, et al. The diagnostic challenge of progressive pseudorheumatoid dysplasia (PPRD): A review of clinical features, radiographic features, and WISP3 mutations in 63 affected individuals. Am J Med Genet C Semin Med Genet 2012;160C:217-29. [Google Scholar | PubMed]
- 3.Hurvitz JR, Suwairi WM, Van Hul W, El-Shanti H, Superti-Furga A, Roudier J, et al. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nat Genet 1999;23:94-8. [Google Scholar | PubMed]
- 4.Pomeranz C, Reid J, Progressive pseudorheumatoid dysplasia: A report of three cases and a review of radiographic and magnetic resonance imaging findings. Skeletal Radiol 2019;48:1323-8. [Google Scholar | PubMed]
- 5.Maatallah K, Boussaa H, Ferjani HL, Kaffel D, Hamdi W. Progressive pseudorheumatoid dysplasia: A rare entity mimicking juvenile idiopathic arthritis. Clin Case Rep 2021;9:e04670. [Google Scholar | PubMed]
- 6.Cefle A, Cefle K, Tunaci M, Ozturk S, Palanduz S. A case of progressive pseudorheumatoid arthropathy of “childhood” with the diagnosis delayed to the fifth decade. Int J Clin Pract 2006;60:1306-9. [Google Scholar | PubMed]
- 7.Taspinar O, Kelesoglu F, Keskin Y, Uludag M. Progressive pseudorheumatoid dysplasia misdiagnosed as seronegative juvenile idiopathic arthritis. Ethiop J Health Sci 2016;26:397-400. [Google Scholar | PubMed]
- 8.Ehl S, Uhl M, Berner R, Bonafé L, Superti-Furga A, Kirchhoff A. Clinical, radiographic, and genetic diagnosis of progressive pseudorheumatoid dysplasia in a patient with severe polyarthropathy. Rheumatol Int 2004;24:53-6. [Google Scholar | PubMed]
- 9.Lachman RS. International nomenclature and classification of the osteochondrodysplasie (1997). Pediatr Radiol 1998;28:737-44. [Google Scholar | PubMed]
- 10.Oestreich AE. Mega os trigonum in progressive pseudorheumatoid dysplasia. Pediatr Radiol 2002;32:46-8. [Google Scholar | PubMed]
- 11.Yang X, Song Y, Kong Q. Diagnosis and surgical treatment of progressive pseudorheumatoid dysplasia in an adult with severe spinal disorders and polyarthropathy. Joint Bone Spine 2013;80:650-2. [Google Scholar | PubMed]
- 12.Bezalel T, Carmeli E, Been E, Kalichman L. Scheuermann’s disease: Current diagnosis and treatment approach. J Back Musculoskelet Rehabil 2014;27:383-90. [Google Scholar | PubMed]
- 13.Gao YS, Ding H, Zhang CQ. Total hip arthroplasty in a 17-year-old girl with progressive pseudorheumatoid dysplasia. J Clin Rheumatol 2013;19:138-41. [Google Scholar | PubMed]






Related Articles in Journal of Orthopaedic Case Reports
 January 1, 2025 Custom-Made 3D-Printed Augments and Cages: An Effective Solution for Managing Severe Acetabular Bone Loss
January 1, 2025 Custom-Made 3D-Printed Augments and Cages: An Effective Solution for Managing Severe Acetabular Bone Loss October 1, 2025 Retrieval Technique of a Broken Guidewire During Core Decompression: Bailout Options – A Case Report
October 1, 2025 Retrieval Technique of a Broken Guidewire During Core Decompression: Bailout Options – A Case Report August 1, 2026 Functional and Cosmetic Outcome in Vertical Midline versus Horizontal Curvilinear Patella-Centered Incision in Internal Fixation of Patellar Fractures
August 1, 2026 Functional and Cosmetic Outcome in Vertical Midline versus Horizontal Curvilinear Patella-Centered Incision in Internal Fixation of Patellar Fractures March 1, 2026 Acute Cauda Equina Syndrome Due to Primary Lumbar Spinal Hydatid Cyst: A Rare Case
March 1, 2026 Acute Cauda Equina Syndrome Due to Primary Lumbar Spinal Hydatid Cyst: A Rare Case