
This experience illustrates the importance of keeping a less common diagnosis as a differential diagnosis. Histopathological examination plays a vital role in diaphyseal bone lesion in infants, as Unifocal LCH can mimic osteomyelitis or malignancy.
Dr. Kavin Sathish, Department of Orthopaedic Surgery, Kauvery Hospitals, No-6, Royal Road, Cantonment, Tiruchirapalli, Tamil Nadu, India. E-mail: sathishkavin525@gmail.com
Abstract
Introduction: Langerhans cell histiocytosis (LCH) is a group of idiopathic disorders characterized by proliferation of bone marrow-derived Langerhans cells. It has a multitude of presentations – ranging from unifocal single-system disorder to multisystem disorder. Most of these present in childhood; however, rare cases have been reported in adulthood as well.
Case Report: Herein, an 8-month-old boy presented with refusal to weight-bear on the right lower limb for 1 week. Mild elevation of C-reactive protein and white blood cell count. A plain radiograph showed a lytic lesion in the femur with a periosteal reaction. Magnetic resonance imaging showed a subperiosteal fluid collection suggestive of muscle inflammation. The patient was taken for curettage and biopsy of the lesion. Histopathology confirmed LCH, with positive for S100 (histiolytic marker) and CD1a (Langerhan cell-specific marker). Whole body positron emission tomography/ Computed tomography confirmed a solitary lesion consistent with single system. Unifocal bone LCH. Systemic therapy was initiated with the help of pediatric hematologist. Clinical and radiological outcomes are observed periodically. The patient was initially evaluated for osteomyelitis. However, we arrived at a diagnosis of LCH on the grounds of morphology and immunohistochemistry.
Conclusion: This case highlights the importance of biopsy for diagnosing bone lesions in infants, as unifocal LCH can mimic osteomyelitis or malignancy. We reported a case of diaphyseal femur LCH in an infant, which has not been reported in the Indian literature until now.
Keywords: Langerhans cell histiocytosis, femur, Infant, solitary bone lesion.
Langerhans cell histiocytosis (LCH) was formerly known as histiocytosis X, as the lesional cells were initially unknown. LCH demonstrates neoplastic proliferation of histiocytes and other inflammatory cells, leading to accumulation and pathological dissemination of histiocytes, resulting in destruction of hard and soft tissues. The diagnosis of LCH is solely based on microscopic examination, and the disease is broadly categorised into three disorders based on the clinical presentations, namely, Hand-Schüller-Christian disease, Letterer-Siwe disease and eosinophilic granuloma [1]. LCH is the most common pediatric histiocytic disorder, with an incidence of four to eight per million children. Although LCH can present at any age, it is primarily a disease of childhood, with a median age of 3–4 years, and has been reported even in infants under 1 year old. The disease ranges from unifocal single-system to life-threatening multisystem involvement [2].
The skeleton is the most commonly affected site in pediatric LCH, with bone lesions occurring in up to 80% of cases. [3] A solitary bone lesion, historically referred to as ‘eosinophilic granuloma’, represents single-system LCH. Frequently involved bones are the skull, pelvis, ribs and long bones such as the femur. These lesions are often lytic with possible periosteal reactions and can resemble osteomyelitis or primary malignant bone tumours on imaging [4,5]. The purpose of reporting this article is to discuss the importance of integrating clinical, radiological and morphological features in the diagnosis of LCH. Management and prognosis of LCH are based on the clinical presentation of the lesion.
An 8-month-old well-nourished boy was brought to our hospital with refusal to bear weight on his right leg for 1 week. There was no history of trauma, fever, or weight loss. General physical examination was normal. Local examination revealed mild tenderness over the swelling on the right thigh. The distal neurovascular status of the limb was intact. Haematological investigations showed a white blood cell count of 20810 cells/cumm and a C-reactive protein level of 20.7 mg/dL; these values are suggestive of acute osteomyelitis. An X-ray of the right femur revealed a punched-out lytic lesion with a periosteal reaction over the shaft region, a feature suggestive of an aggressive bone lesion (Fig. 1).
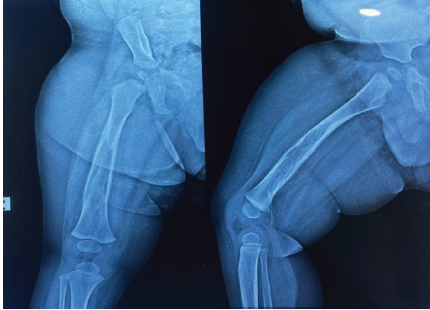
Figure 1: Pre-operative X-ray – Diaphyseal lytic lesion with cortical thickening and periosteal reaction.
Magnetic resonance imaging of the right femur revealed cortical thickening, focal cortical defect, periosteal reaction and surrounding fluid collection suggestive of osteomyelitis (Fig. 2).
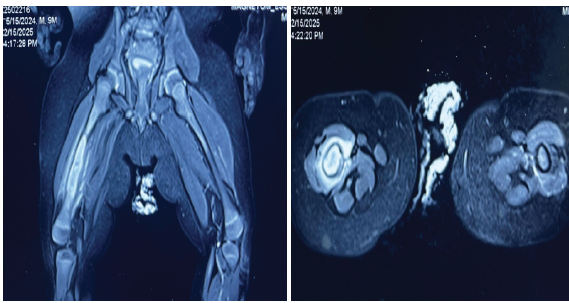
Figure 2: Magnetic resonance imaging – cortical thickening with focal cortical defect, periosteal reaction with surrounding fluid collection T2 cuts – Coronal image and Axial image.
Radiologically, osteomyelitis was given as a possibility, with all investigations leading to a clinical diagnosis of osteomyelitis. The patient was taken for incision and drainage and curettage of the lesion. Intraoperatively, under C-arm guidance, a lytic lesion was identified, and curettage was done; serous discharge was observed, and slough tissue along with curetted material from the femur canal was sent for histopathology (Figs. 3 and 4).
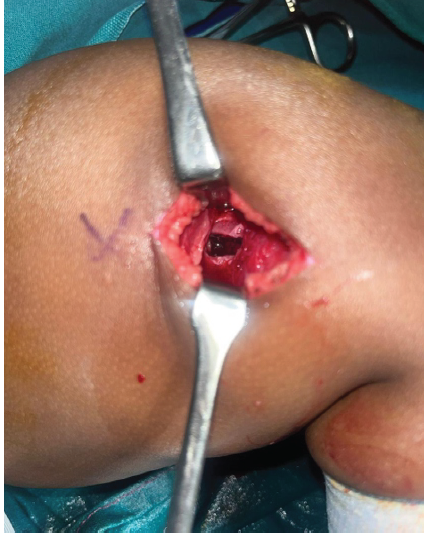
Figure 3: Intraoperative image – window made through cortical defect, curettage and thorough washout given.
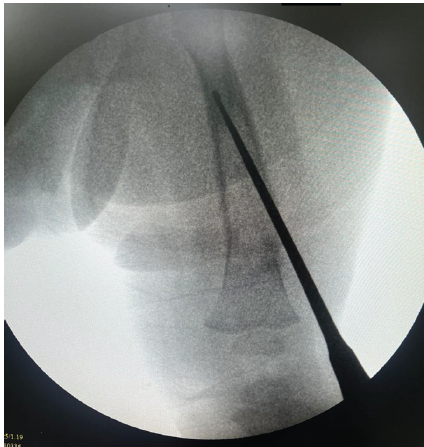
Figure 4: C-arm guidance femur canal curettage done.
The specimen revealed a proliferation of sheets of large mononuclear pale-staining cells with ill-defined cellular margins (histiocytes), interspersed with inflammatory cells. These histiocytes had cleaved/grooved nuclei with vesicular chromatin and small nucleoli. The inflammatory infiltrate was predominantly composed of eosinophils. These findings were histopathologically suggestive of LCH (Fig. 5).
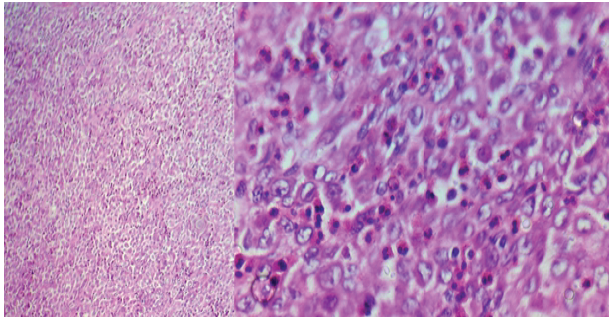
Figure 5: (a) Sheets of histiocytes and chronic inflammatory infiltrate hematoxylin and eosin (H&E), ×100. (b) Shows sheets of Langerhans cells with grooved/cleaved coffee bean nuclei admixed with numerous eosinophils H&E, ×400.
To confirm this, immunohistochemical staining was performed; the neoplastic cells were positive for S100 (histiocytic marker) and CD1a (Langerhans cell-specific marker) (Fig. 6).
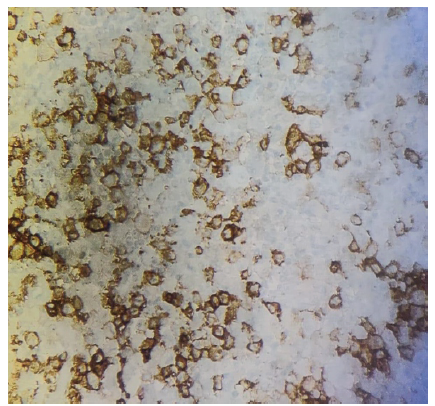
Figure 6: Langerin immunohistochemistry section shows large histiocytic cells with positive membranous staining for CD1a.
Positron emission tomography-computed tomography revealed no other lesions elsewhere. Based on the age of the patient and clinical presentation of the lesion involving a single bone uni-system, a unifocal type of LCH was made (Fig. 7).
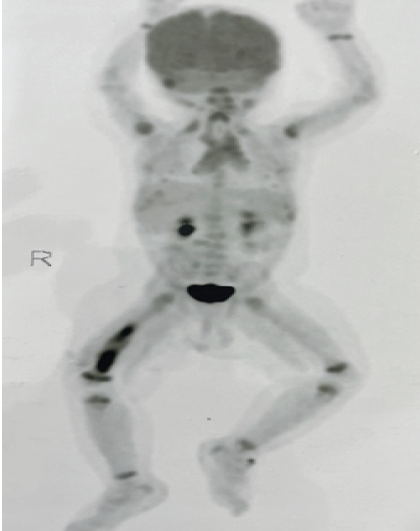
Figure 7: Positron emission tomography computed tomography – shows no other systemic involvement.
The patient was started on systemic chemotherapy by a paediatric haematologist for a period of 6 months. Clinical and radiological outcomes were observed at 6 weeks, 3 months, 6 months, and 9 months of duration. The patient is doing well without progression of disease (Figs 8 and 9).
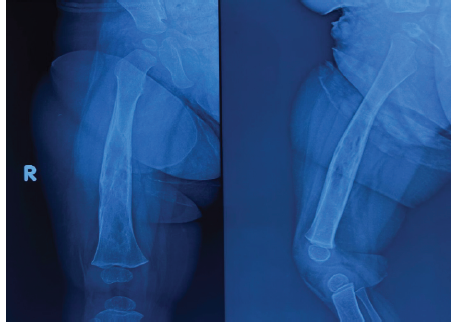
Figure 8: 6 weeks post-operative X-ray.
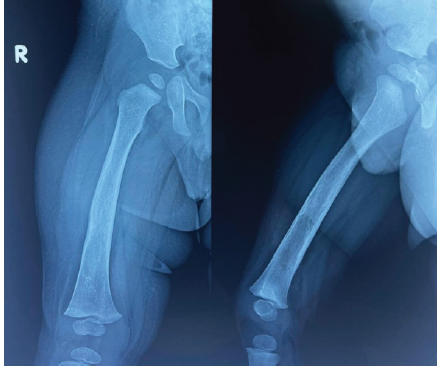
Figure 9: 6 months post-operative X-ray.
LCH is currently regarded as a myeloid/histiocytic neoplasm, with a remarkably broad clinical spectrum, ranging from isolated skin or bone lesions to a disseminated disease that can involve nearly any organ. LCH is generally regarded as a sporadic disease that occurs predominantly in the pediatric population [6]. The reported incidence of LCH ranges from 2.6 to 8.9 cases/million children younger than 15 years/year, with a median age at diagnosis of 3 years [7]. Our case was an infant of 8 months of age. The exact incidence of LCH in adults is much less defined: The only available data are for disseminated disease, with 0.07 cases/million/year [7,8]. This distinct group of diseases that is collectively referred to as LCH is categorized into three variants based on age and clinical presentation; clear separation is difficult on the basis of histological manifestations. These variants include (1) the acute disseminated form with multiple system involvement, often occurring mainly in infants (Letterer-Siwe disease). (2) Chronic disseminated form with osseous lesions, which are frequently multiple and with extraskeletal lesions (Hand-Schuller-Christian disease). (3) Chronic localised form with solitary or multiple skeletal lesions and occasional extraskeletal involvement, mainly seen in adults (eosinophilic granuloma). Hashimoto-Pritzker syndrome is a congenital form of LCH presenting with deep subcutaneous skin lesions [8,9]. In recent times, all these terminologies have become obsolete and have been replaced by ‘unisystem disease’ or ‘multisystem disease’. Our child presented with isolated bone disease, and extensive radiological workup did not reveal any other lesion. [10,11] In all age groups, isolated diaphyseal destruction of the long bone with fusiform periosteal reaction and extensive peripheral oedema, vertebra plana of the spine, and bevelled-edge skull defects accompanied by soft tissue masses strongly suggest an LCH diagnosis. Moreover, the multiple bone osteolytic destruction in children and adolescents strongly suggests an LCH diagnosis [12]. There are 2 main broad theories about its pathogenesis: one is that it is a reactive process due to inappropriate immune stimulation, and the other is that it is a neoplastic disorder. In 2010, Badalian-Very et al. described abnormal CD1a+CD207+histiocytes that carried a somatic variant of the BRAFV600E oncogene in 57% of 61 affected patients. This variant has been confirmed in numerous cohorts, such as a paediatric case series in France that included 315 patients, with detection of BRAFV600E in 54.6%. Other activating pathogenic variants have been found in the RAS-RAF-MEK-ERK pathway involved in the proliferation, survival, differentiation and activation of myeloid dendritic cell precursors [13]. A definitive diagnosis of LCH requires a combination of clinical presentation, histology, and immunohistochemistry. The inflammatory infiltrate contains various proportions of LCH cells, the disease hallmark, which are round and have characteristic “coffee-bean” cleaved nuclei and eosinophilic cytoplasm. Although a well-defined histologically characteristic appearance of the LCH lesions on haematoxylin and eosin-stained sections is present, positive CD1a and/or CD207 (Langerin) staining of the lesional cells is required for a definitive diagnosis [13,14]. The preferred first-line therapy for multisystem LCH is the vinblastine-prednisone regimen. We treated our patient using a vinblastine and methylprednisone regimen for 6 months. Methotrexate and hydroxyurea have been investigated as low-toxicity oral alternatives in LCH. BRAF inhibitors such as vemurafenib demonstrated a 41% overall response rate in refractory cases [14].
LCH is a rare disease with a variable presentation, which can make the diagnosis challenging. Unifocal single-system bone LCH in infants should remain a key differential consideration for lytic bone lesions that mimic infection or malignancy. In this case, prompt histological confirmation during the initial procedure enabled accurate diagnosis and appropriate management. With proper follow-up and treatment, a unifocal unisystem disease has a good prognosis as compared to a multisystem disease. We have reported single-system diaphyseal LCH in an infant’s femur not yet published in Indian literature.
This case highlights the importance of biopsy for diagnosing bone lesions in infants, as unifocal LCH can mimic osteomyelitis or malignancy. This experience illustrates the importance of keeping a less common diagnosis as a differential diagnosis.
References
- 1. Madrigal-Martínez-Pereda C, Guerrero-Rodríguez V, Guisado-Moya B, Meniz-García C. Langerhans cell histiocytosis: Literature review and descriptive analysis of oral manifestations. Med Oral Patol Oral Cir Bucal. 2009;14:E222–8. [Google Scholar] [PubMed]
- 2. Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. N Engl J Med. 2018 Aug 30;379(9):856-868. doi: 10.1056 [Google Scholar] [PubMed]
- 3. Demellawy, Dina El et al. Langerhans cell histiocytosis: a comprehensive review. Pathology, Volume 47, Issue 4, 294 – 301 [Google Scholar] [PubMed]
- 4. Stålemark H, Laurencikas E, Karis J, Gavhed D, Fadeel B, Henter JI. Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr Blood Cancer. 2008 Jul;51(1):76-81. doi: 10.1002/pbc.21504. [Google Scholar] [PubMed] [CrossRef]
- 5. Baumgartner I, von Hochstetter A, Baumert B, Luetolf U, Follath F. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997 Jan;28(1):9-14. doi: 10.1002/(sici)1096-911x(199701)28:1<9::aid-mpo3>3.0.co;2-p. [Google Scholar] [PubMed] [CrossRef]
- 6. Goyal G, Shah MV, Hook CC, Wolanskyj AP, Call TG, Rech KL, Go RS. Adult disseminated Langerhans cell histiocytosis: incidence, racial disparities and long-term outcomes. Br J Haematol. 2018 Aug;182(4):579-581. doi: 10.1111/bjh.14818 [Google Scholar] [PubMed] [CrossRef]
- 7. Yashoda-Devi B, Rakesh N, Agarwal M. Langerhans cell histiocytosis with oral manifestations: A rare and unusual case report. J Clin Exp Dent. 2012;4:e252–5. doi: 10.4317/jced.50728 [Google Scholar] [PubMed] [CrossRef]
- 8. Aruna DR, Pushpalatha G, Galgali S, Prashanthy Langerhans cell histiocytosis. J Indian Soc Periodontol. 2011;15:276–9. doi: 10.4103/0972-124X.85675. [Google Scholar] [PubMed] [CrossRef]
- 9. Zhao M, Tang L, Sun S, Cui J, Chen H. Radiologic findings that aid in the reduction of misdiagnoses of Langerhans cell histiocytosis of the bone: a retrospective study. World J Surg Oncol. 2021 May 10;19(1):146. doi: 10.1186/s12957-021-02261-y. [Google Scholar] [PubMed] [CrossRef]
- 10. Astigarraga I, García-Obregón S, Pérez-Martínez A, Gutiérrez-Carrasco I, Santa-María V, Rodríguez-Vigil Iturrate C, et al. Histiocitosis de células de Langerhans. Avances en la patogenia y práctica clínica. An Pediatr (Barc). 2022;97:130. [Google Scholar] [PubMed]
- 11. Krooks, Jolie et al. Langerhans cell histiocytosis in children Journal of the American Academy of Dermatology, Volume 78, Issue 6, 1047 – 1056 [Google Scholar] [PubMed]
- 12. Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, Egeler RM, Janka G, Micic D, Rodriguez-Galindo C, Van Gool S, Visser J, Weitzman S, Donadieu J; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013 Feb;60(2):175-84. doi: 10.1002/pbc.24367. [Google Scholar] [PubMed] [CrossRef]
- 13. Hamoud, M., Alchaikh Hassan, R., & Dasanu, C. A. (2025). Selecting optimal therapy for Langerhans cell histiocytosis: current state and future directions. Expert Opinion on Pharmacotherapy, 1–4. [Google Scholar] [PubMed]
- 14. Feng S, Han L, Yue M, Zhong D, Cao J, Guo Y, Sun Y, Zhang H, Cao Z, Cui X, Liu R. Frequency detection of BRAF V600E mutation in a cohort of pediatric langerhans cell histiocytosis patients by next-generation sequencing. Orphanet Journal of Rare Diseases. 2021 Jun 11;16(1):272. [Google Scholar] [PubMed]
- 15. Chang, W.F., Hsu, Y.C., Wu, Y.D., Kuo, C.L. and Huang, G.S., 2016. Localized Langerhans cell histiocytosis masquerading as Brodie’s abscess in a 2-year-old child: a case report. EXCLI journal, 15, p.33. [Google Scholar] [PubMed]




Related Articles in Journal of Orthopaedic Case Reports
 June 1, 2026 Hydatid Disease Masquerading as Infected Non-Union of Subtrochanteric Femur: A Rare Case Report
June 1, 2026 Hydatid Disease Masquerading as Infected Non-Union of Subtrochanteric Femur: A Rare Case Report May 1, 2026 Idiopathic Congenital Dislocation of the Knee: Case Series of Spontaneous Reduction
May 1, 2026 Idiopathic Congenital Dislocation of the Knee: Case Series of Spontaneous Reduction April 1, 2026 Incidental Intramedullary Lipoma of the Proximal Femur Detected Following Trauma: A Case Report
April 1, 2026 Incidental Intramedullary Lipoma of the Proximal Femur Detected Following Trauma: A Case Report November 1, 2025 Incidence and Functional Impact of Malrotation after Intramedullary Nailing of Femoral Shaft Fractures: A Prospective Computed Tomography-based Observational Study
November 1, 2025 Incidence and Functional Impact of Malrotation after Intramedullary Nailing of Femoral Shaft Fractures: A Prospective Computed Tomography-based Observational Study