
While most lipoblastomas are diagnosed in infancy, they can occur in adolescents.
Dr. Lorena V Floccari, Department of Orthopedic Surgery and Sports Medicine, Akron Children’s Hospital, 1 Perkins Sq., Akron, Ohio, 44308, USA. E-mail: lfloccari@akronchildrens.org
Abstract
Introduction: Lipoblastomas are fatty tumours of mesenchymal origin with a peak incidence between the ages of 5 and 6 years. Several cases of lipoblastomas have been reported in the paediatric population, with few in the paraspinal musculature. We present a case of a paraspinal lipoblastoma in an adolescent.
Case Report: An 11-year-old male presented with an asymptomatic thoracic mass. Imaging revealed a 5 cm³ lesion involving the left fourth and fifth ribs with extension into the T4-T5 neural foramen. Biopsy confirmed lipoblastoma. Marginal resection was performed without complication, and there was no recurrence at 2 years.
Conclusion: Lipoblastomas are rare mesenchymal tumours whose differential diagnosis includes lipomas and liposarcomas. While benign, lipoblastomas grow rapidly and can cause a mass effect on neurovascular structures. Most cases are diagnosed in infancy; however, this patient was diagnosed in adolescence.
Keywords: Lipoblastoma, adolescent, differential diagnosis.
Lipoblastomas are fatty tumours of mesenchymal origin that typically occur in the first decade of life [1]. Their peak incidence is between the ages of 5 and 6, with a male-to-female predominance of 4:1 [2]. Although more commonly found in the extremities, these tumours can occur adjacent to the axial skeleton [1,2]. Proximity to the spine can result in a mass effect on neurovascular structures and possible compromise. A radiographic analysis study showed that of paraspinal cases, 38% involved the neural foramina or central canal [3]. Several cases of lipoblastomas have been reported in the paediatric population, with few in the paraspinal musculature [2,4,5,6,7]. We present a case of a paraspinal lipoblastoma in an adolescent.
The patient is an 11-year-old male who initially presented to his paediatrician due to concern for a left upper thoracic hump at the age of 9 years. Radiographs were obtained and reported as thoracic dextroscoliosis measuring 12° without any vertebral body anomalies or paraspinal processes. Given that the patient was neurologically intact and asymptomatic, he was treated conservatively by his paediatrician with observation. The patient then presented 2 years later with cough, lymphopenia, and 6 weeks of fatigue. Infectious work-up included a chest radiograph, which revealed thinning of the left fourth and fifth ribs with an associated calcified mass. Thoracic spine radiographs were obtained with a marker placed within the mass (Fig. 1), which confirmed radiographic and clinical relationships.
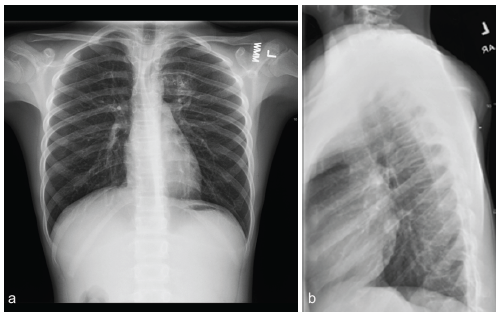
Figure 1: Thoracic spine radiographs (a: Anteroposterior, b: Lateral) performed with an external marker placed on the posterior prominence, demonstrating a calcific-appearing, radiopaque posterior chest wall lesion with 4th and 5th rib thinning and deformity.
Computed tomography and magnetic resonance imaging (MRI) scans of the chest revealed a large heterogeneous, lobulated, and well-defined mass in the posterior mediastinum and paraspinal musculature with extension into the T4-T5 neural foramen. There were both fatty and calcified components. The mass measured approximately 4.6 × 5.2 × 5.0 cm in the anteroposterior, coronal, and craniocaudal dimensions, respectively (Figs. 2 and 3).
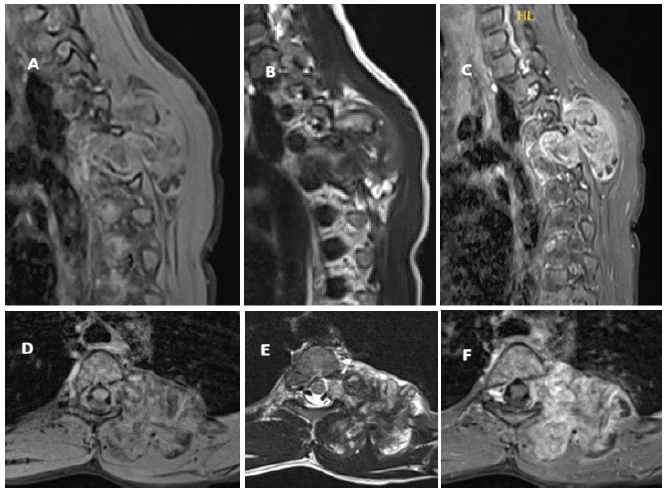
Figure 2: Representative magnetic resonance imaging images, including sagittal (a: T1-weighted, b: T2-weighted, and c: Post-contrast) and axial (d: T1-weighted, e: T2-weighted, and f: Post-contrast) views depicting the calcified T4-T5 lesion with extension into the left neural foramen.
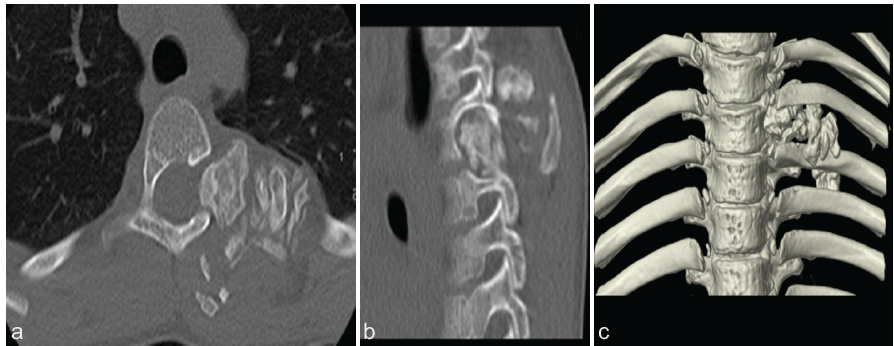
Figure 3: Computed tomography images, including (a) axial, (b) sagittal, and (c) 3D reconstruction depicting a calcified mass with extension into the left neural foramen.
On clinical examination, the patient was 147 cm tall and weighed 30 kg. He was well-developed and well-nourished. He did not have any reported limitations with activity, neurologic symptoms, or bowel or bladder dysfunction. His family history was negative for tumours and positive for scoliosis in his father. On exam, gait was normal with plantigrade feet and no deformity. There was no evidence of spinal dysraphism. On both upright and forward-bending visual inspections of the spine and thorax, there was no spinal rotational deformity, but he did have a palpable non-tender mass in the left upper back, which was soft and fixed in position. He reported no pain, numbness, or radicular symptoms. He had 5/5 strength, sensation was intact to all dermatomes, and his reflexes were normal. To obtain a diagnosis, the patient underwent an ultrasound-guided, core-needle biopsy of the mass with interventional radiology. Twelve samples were sent for pathology. Results were consistent with lipoblastoma, and orthopaedic oncology, paediatric spine surgery, and paediatric general surgery recommended marginal resection of the mass due to proximity to the spinal canal. The family and patient elected to proceed with surgery. The patient underwent general anaesthesia, and neuromonitoring was used. He was placed prone on a spine board and prepped in routine sterile fashion. An oblique incision was made caudal to the left scapula and over the mass. Dissection was performed, revealing two lobes of the tumour. The fourth and fifth ribs were osteotomised approximately 1.5 cm distal to the lateral margin of the tumour, and the costotransverse joints were disarticulated, allowing for the tumour to be removed en bloc with careful dissection from the left T4-T5 neural foramen with no cerebrospinal fluid leakage encountered. Complete excision was performed, haemostasis was achieved, and the pleural defect was repaired. The posterior chest was reconstructed by the general surgery team with placement of a 5 × 6 cm Gore-Tex patch over the posterior chest wall defect. The wound was irrigated, layered closure was performed, and intrathecal morphine was injected into the lumbar interspace. Neuromonitoring was stable throughout the procedure, and the patient was awakened and taken to the post-operative holding area without complication. The final pathology of the tumour confirmed lipoblastoma, and margins were clean (Fig. 4).
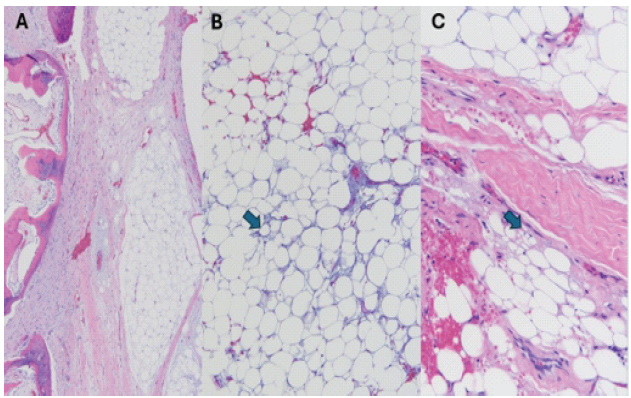
Figure 4: Hematoxylin and eosin-stained sections following decalcification demonstrate lipoblastoma. (a) Lobulated maturing and immature adipose components with myxoid areas, diagnostic of lipoblastoma (×4). The left side of panel A includes areas of the tumor that are ossifying, correlating to the intratumoral calcification seen on imaging. (b) Focal areas of the tumor show lipoblastic features, including signet ring cells (arrow) (×10). (c) Multivacuolated cells with a signet ring appearance (arrow) in another area of the tumor (×20).
The patient followed up at approximately 2 weeks, 6 weeks, 1 year, 2 years, 3 years, and 4 years. Over that time, the patient healed appropriately and was without complication. Each year, the patient undergoes a thoracic spine MRI with and without contrast for surveillance. He developed mild scoliosis (Fig. 5) that required no treatment and was stable 3.5 years post-operatively. There has been no evidence of recurrence.
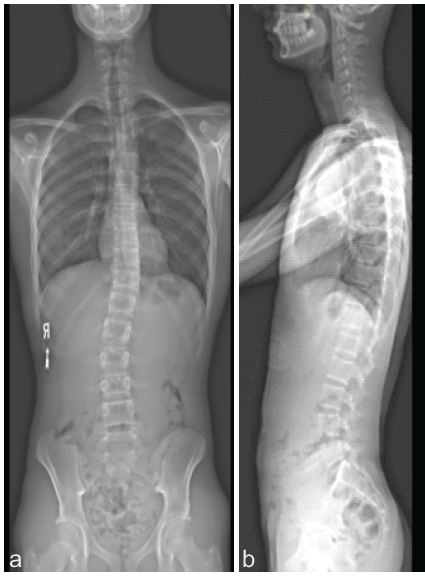
Figure 5: Radiographs (a: Anteroposterior, b: Lateral) obtained 3 years post-operatively demonstrate mild scoliosis but no evidence of recurrence.
Lipoblastoma, the isolated and circumscribed version of lipoblastomatosis, is considered a tumour of infancy [1]. Presentation, physical examination, and advanced imaging do not reliably distinguish lipoblastoma, lipoma, and liposarcoma. Therefore, a biopsy is required to obtain a diagnosis. One key reason diagnosis based on imaging is challenging is that calcifications often present within fatty tumours secondary to fat necrosis. Dystrophic calcifications accumulate in dead or necrotic fat tissue and appear on imaging studies. Radiographic studies have reported that, while calcifications are not specific, they are more likely to appear in malignant adipocytes. The studies found calcifications in 32% of liposarcomas and 11% of lipomas [8,9]. Additional factors that were found to be significantly associated with liposarcomas included size >10 cm, presence of thick septa, presence of globular and/or nodular non-adipose areas, and lesions of <75% fat [8]. Histologically, the key to diagnosis is the presence of lipoblasts at different stages of development that range from spindle-shaped pre-adipocytes to a mature fat cell displaying the classic “signet ring” pattern [1]. Recent studies have shown that abnormalities on chromosome 8 and rearrangements of the PLAG1 gene may be related to lipoblastoma and lipoblastomatosis [10]. The prognosis for lipoblastoma following marginal resection is excellent, with a reported rate of local recurrences ranging from 9% to 22% [11]. If recurrence does occur, it is more commonly seen in infiltrative lipoblastomatosis. The most important factors related to recurrence have been shown to be incomplete resection and the presence of diffuse disease rather than any morphologic feature [1].
Evaluation for pediatric spinal pathology is routine for pediatricians. While adolescent idiopathic scoliosis is relatively common, it is important to consider a broad differential when a patient presents with spinal asymmetry. Our study demonstrates a rare tumor in a unique age group. At 4-year follow-up, there is evidence that treatment of lipoblastoma with marginal resection and surveillance is applicable to adolescent patients. As seen in the patient discussed, a multidisciplinary approach is often required when the mass is adjacent to critical structures in the chest and spine. There is no proven role for adjuvant chemotherapy or radiation therapy. Follow-up is centered around standard post-operative wound healing evaluation and care. Surveillance is a yearly MRI of the post-operative site to evaluate for recurrence.
Our report suggests that while lipoblastoma in an adolescent is exceedingly rare, the work-up and treatment can be the same as that of lipoblastomas found in all age groups.
References
- 1. Weiss SW, Goldblum JR, Enzinger FM. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia, PA, Pennsylvania: Mosby Elsevier; 2008. p. 452-5. [Google Scholar] [PubMed]
- 2. Seroto P, Kelly A, Sadiki T, Younus A. Paraspinal lipoblastoma with multidirectional spread occurring in a pre-school child. Interdiscip Neurosurg 2019;18:100503. [Google Scholar] [PubMed]
- 3. Degnan AJ, Jelinek JS, Murphey MD. Lipoblastoma: Computed tomographic and magnetic resonance imaging features correlate with tumor behavior and pathology. Pediatr Radiol 2021;51:614-21. [Google Scholar] [PubMed]
- 4. Gupta G, Garg R, Wadhwa C, Bansal H, Kaushal RK. A rare primary dumbbell lipoblastoma. Asian J Neurosurg 2018;13:83-5. [Google Scholar] [PubMed]
- 5. Brodsky JR, Kim DY, Jiang Z. Cervical lipoblastoma: Case report, review of literature, and genetic analysis. Head Neck 2007;29:1055-60. [Google Scholar] [PubMed]
- 6. Saga M, Yamaura A, Miyagawa T. Lipoblastomatosis extended into the lumbar spinal canal in a child: A case report. Pediatr Neurosurg 2023;58:168-72. [Google Scholar] [PubMed]
- 7. Giri SA, Diyora B, Giri D, Sharma A. Primary spinal extradural lipoblastoma: Rare occurrence. J Pediatr Neurosci 2014;9:300-1. [Google Scholar] [PubMed]
- 8. Kransdorf MJ, Bancroft LW, Peterson JJ, Murphey MD, Foster WC, Temple HT. Imaging of fatty tumors: Distinction of lipoma and well-differentiated liposarcoma. Radiology 2002;224:99-104. [Google Scholar] [PubMed]
- 9. Peterson JJ, Kransdorf MJ, Bancroft LW, O’Connor MI. Malignant fatty tumors: Classification, clinical course, imaging appearance and treatment. Skeletal Radiol 2003;32:493-503. [Google Scholar] [PubMed]
- 10. Gisselsson D, Hibbard MK, Dal Cin P, Sciot R, Hsi BL, Kozakewich HP, et al. PLAG1 alterations in lipoblastoma: Involvement in varied mesenchymal cell types and evidence for alternative oncogenic mechanisms. Am J Pathol 2001;159:955-62. [Google Scholar] [PubMed]
- 11. Mwntzel T, Calonje E, Fletcher CD. Lipoblastoma and lipoblastomatosis: A clinicopathological study of 14 cases. Histopathology 1993;23:527-33. [Google Scholar] [PubMed]




Related Articles in Journal of Orthopaedic Case Reports
 August 1, 2026 Beyond Pipkin IV: Management of Femoral Head-neck Fracture-dislocation with Posterior Acetabular Wall and Peritrochanteric Fracture in an Adolescent Using a Modified Kocher-Langenbeck Approach
August 1, 2026 Beyond Pipkin IV: Management of Femoral Head-neck Fracture-dislocation with Posterior Acetabular Wall and Peritrochanteric Fracture in an Adolescent Using a Modified Kocher-Langenbeck Approach May 1, 2026 Atypical Hip Pain due to Intra-articular Osteoid Osteoma Treated by Surgical Hip Dislocation in an Adolescent: Case Report
May 1, 2026 Atypical Hip Pain due to Intra-articular Osteoid Osteoma Treated by Surgical Hip Dislocation in an Adolescent: Case Report May 1, 2026 Reconstructive Surgery for Foot Ectrodactyly in an Adolescent Patient: A Case Report
May 1, 2026 Reconstructive Surgery for Foot Ectrodactyly in an Adolescent Patient: A Case Report March 1, 2026 Single-Stage Treatment of a Patellar Osteochondral Defect in an Adolescent Using Autologous Minced Cartilage – Platelet-Rich Plasma Scaffold Technique: A Case Report and Technical Considerations
March 1, 2026 Single-Stage Treatment of a Patellar Osteochondral Defect in an Adolescent Using Autologous Minced Cartilage – Platelet-Rich Plasma Scaffold Technique: A Case Report and Technical Considerations