
In severe Ollier disease, rapid enlargement of a giant lesion may clinically mimic malignant transformation; diagnosis and management should rely on integrated clinicoradiological assessment, selective biopsy of suspicious lesions, and structured long-term surveillance.
Dr. Kshitij Pillai, Department of Orthopaedics, JSS Medical College, JSS Hospital, Mahatma Gandhi Road, Mysuru - 570 004, Karnataka, India. E-mail: kshitijpillai7@gmail.com
Abstract
Introduction: Ollier disease is a rare sporadic enchondromatosis characterised by multiple asymmetrically distributed enchondromas, progressive skeletal deformity, and risk of malignant transformation.
Case Report: A 16-year-old boy presented with severe childhood-onset multifocal appendicular deformity, short stature, dysmorphic facies, developmental delay, and a giant left proximal humeral lesion that raised concern for secondary chondrosarcoma. The first swelling was noticed over the left hand at about 3 months of age, followed by progressive asymmetric lesions involving the upper limbs, pelvis, femora, tibiae, knees, and hands. Examination showed multiple hard, non-tender bony swellings, restricted shoulder movement, limb-length inequality, angular deformity, and gait disturbance. Serial radiographs demonstrated widespread expansile intramedullary chondroid lesions with cortical expansion and progressive deformity. Because of the interval enlargement of the shoulder lesion, paediatric, genetic, endocrine, and surgical oncology opinions were obtained. Ultrasound-guided core biopsy showed hyaline cartilage without increased cellularity, cytological atypia, or mitotic activity, favouring a benign chondroid lesion. Molecular testing was advised but could not be performed because of financial constraints. The patient was managed with non-steroidal anti-inflammatory drugs, physiotherapy, and surveillance.
Conclusion: At 6 months, he reported symptomatic improvement, and at 1 year, there was no documented clinical progression. This case highlights the diagnostic complexity of severe enchondromatosis, the value of longitudinal imaging review and biopsy of suspicious lesions, and the practical role of multidisciplinary surveillance when advanced molecular testing is not feasible.
Keywords: Ollier disease, enchondromatosis, enchondroma, proximal humerus, chondrosarcoma surveillance.
Ollier disease is a rare, non-hereditary enchondromatosis characterised by multiple intramedullary cartilaginous tumours, classically distributed asymmetrically in the appendicular skeleton. It usually presents in childhood and may cause limb-length discrepancy, angular deformity, pathological fracture, and functional limitation [1,2,3,4,5]. Unlike hereditary multiple osteochondromas, the lesions are intramedullary rather than exophytic; unlike Maffucci syndrome, soft-tissue haemangiomas are absent [1,2,3]. Post-zygotic somatic mosaic mutations in isocitrate dehydrogenase (IDH)1 and IDH2 are recognised as the principal molecular mechanism underlying Ollier disease and Maffucci syndrome, explaining the segmental and asymmetric distribution of lesions [2]. The main lifelong concern is malignant transformation, most often to chondrosarcoma, especially in large, painful, rapidly enlarging lesions or lesions in high-risk sites, such as the pelvis, shoulder girdle, distal femur, and proximal tibia [3,4,5]. We report a severe childhood-onset case most consistent with Ollier disease in an adolescent male with extensive deformity and a giant proximal humeral lesion that clinically suggested malignant transformation but was benign on core biopsy.
A 16-year-old male, the firstborn child of non-consanguineous parents, presented with multiple progressively enlarging bony swellings involving all four limbs. He had completed schooling up to the tenth grade. Birth history was significant for breech presentation, Caesarean delivery, low birth weight (1.8 kg), delayed cry at birth, and neonatal intensive care admission. Macrocephaly was noted at approximately 2.5 months of age, and subsequent evaluation reportedly showed bilateral frontotemporal extra-axial collection, interpreted as subdural hygroma/haematoma, for which a ventriculoperitoneal shunt was placed at approximately 3 months of age. The first musculoskeletal swelling was noticed over the left hand at around 3 months. Over the following years, additional asymmetric swellings developed around the metaphyseal regions of the upper and lower limbs, with progressive bowing, shortening, limb-length discrepancy, difficulty walking, difficulty squatting and sitting cross-legged, and activity-related pain. There was no night pain, constitutional symptom, neurological deficit, or similar illness in first-degree relatives. Examination showed short stature, frontal bossing, hypertelorism, down-slanting palpebral fissures, a broad nasal bridge, a long face, and dental malocclusion. No cutaneous haemangiomas were identified. Multiple hard bony swellings were present over all four limbs. The dominant lesion was a giant globular mass arising from the left shoulder/proximal humerus. Additional swellings were present around both wrists, distal femora, proximal tibiae, distal tibiae, and hands. The swellings were hard, well-defined, fixed to bone, and covered by normal mobile skin. Distal neurovascular status was intact. Bilateral shoulder movement was restricted, especially on the left. Lower-limb malalignment, shortening, and muscle wasting contributed to gait impairment (Fig. 1).
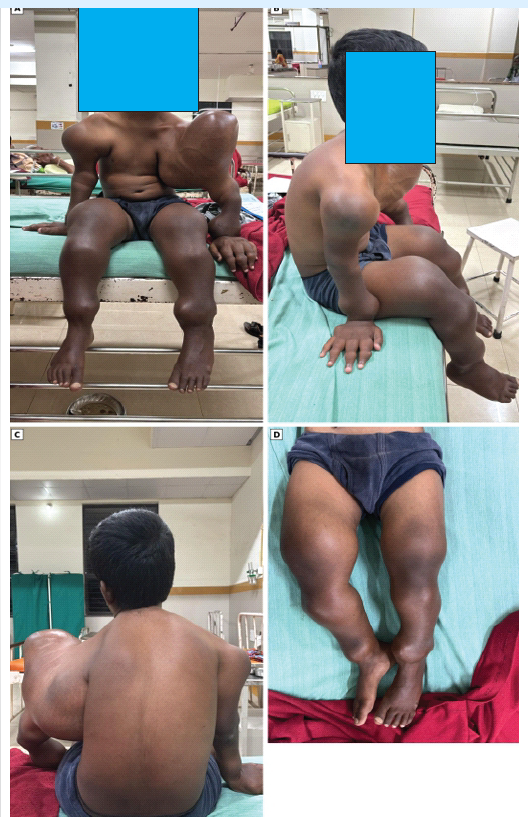
Figure 1: Clinical photographs. (a) Frontal view demonstrating short stature and upper-limb deformity, with facial identifying details concealed, (b) Lateral view showing the giant left shoulder/proximal humeral swelling and upper-limb asymmetry, (c) Posterior view showing marked shoulder-girdle asymmetry, (d) Lower-limb photograph showing bilateral deformity and prominent peri-knee swellings.
Childhood radiographs had been reported as metaphyseal chondrodysplasia; however, retrospective review of serial radiographs was more compatible with enchondromatosis. The films demonstrated multiple expansile intramedullary chondroid lesions with metaphyseal predominance, cortical expansion, and deformity involving both upper limbs, the pelvis and proximal femora, distal femora and proximal tibiae, hands, and distal forearms (Fig. 2).
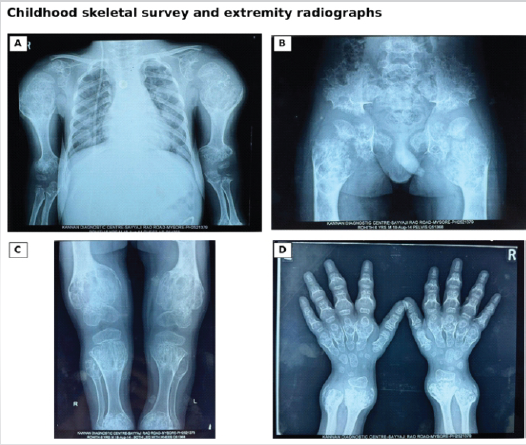
Figure 2: Childhood skeletal survey and extremity radiographs. (a) Bilateral upper-limb and shoulder radiograph showing expansile chondroid lesions, (b) Pelvic radiograph showing bilateral pelvic and proximal femoral involvement, (c) Bilateral knee radiograph showing metaphyseal expansion and deformity, (d) Hand radiograph showing multiple expansile lesions involving the metacarpals, phalanges, and distal forearms.
Adolescent radiographs showed persistent multifocal disease and marked interval enlargement of a giant expansile chondroid lesion centred in the left proximal humerus (Fig. 3).
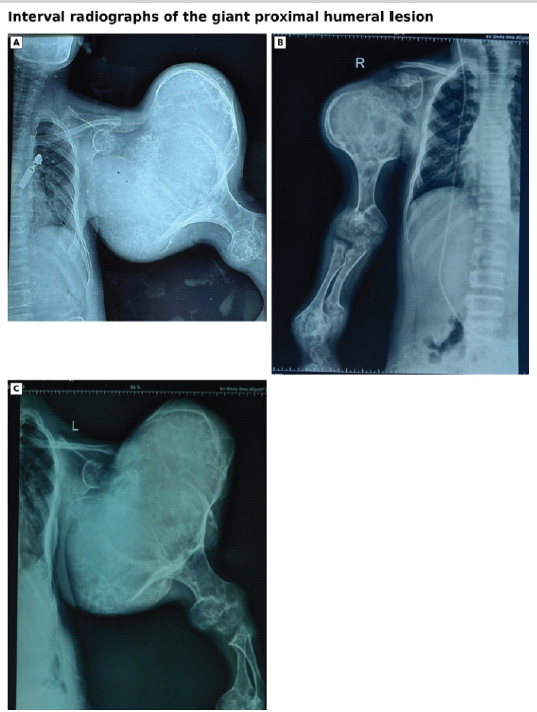
Figure 3: Interval radiographs of the giant proximal humeral lesion. (a and b) Radiograph of left and right shoulder, respectively, showing a large expansile chondroid lesion with proximal humeral deformity. (c) One-year follow-up interval radiograph demonstrating persistent massive enlargement and no increase in size.
No definite permeative destruction or soft-tissue mass was evident on the available radiographs, but the size and enlargement made secondary chondrosarcoma an important concern. Laboratory evaluation showed mild anaemia (haemoglobin 9.8 g/dL) and elevated erythrocyte sedimentation rate (50 mm in the 1st h). Serum calcium and phosphorus were normal; alkaline phosphatase was mildly elevated, vitamin D was suboptimal, parathyroid hormone was normal, viral screening was non-reactive, and previous thyroid function testing was normal. The biochemical profile did not support nutritional rickets as the primary diagnosis. A multidisciplinary review was obtained. Paediatrics documented short stature, developmental delay, and dysmorphic facies and advised endocrine, ophthalmological, inflammatory, haematological, thyroid, biopsy/excision, and genetic evaluation. Surgical oncology recommended image-guided biopsy of the enlarging shoulder lesion. Genetics considered the phenotype most compatible with Ollier disease, counselled the family, and suggested DNA storage with possible IDH1/IDH2 testing on a research basis. Endocrine follow-up was advised for short stature assessment. Ultrasound-guided core biopsy of the left shoulder lesion was performed under local anaesthesia, and three cores were obtained. Histopathology showed fragments of hyaline cartilage without increased cellularity, cytological atypia, or mitotic activity, favouring a benign chondroid lesion (Fig. 4).
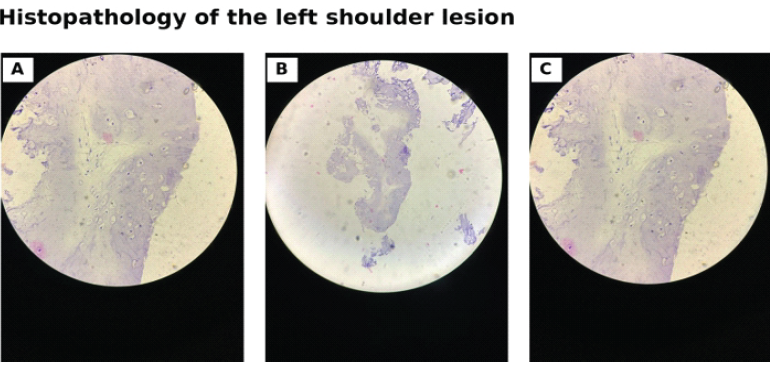
Figure 4: Histopathology of the left shoulder lesion. Photomicrographs of ultrasound-guided core biopsy show fragments of hyaline cartilage without increased cellularity, cytological atypia, or mitotic activity, favoring a benign chondroid lesion.
Video-derived standing and gait stills showed a broad-based, slow, short-step gait with lateral trunk sway, lower-limb malalignment, altered weight transfer, and persistent postural asymmetry (Fig. 5).

Figure 5: Video-derived gait and standing stills. (a and b) Posterior views showing asymmetric shoulder levels, broad-based stance, lower-limb malalignment, and altered weight transfer during gait. (c and d) Anterior views showing short-step progression, lateral trunk sway, and persistent postural asymmetry caused by extensive appendicular deformity and the giant left proximal humeral lesion.
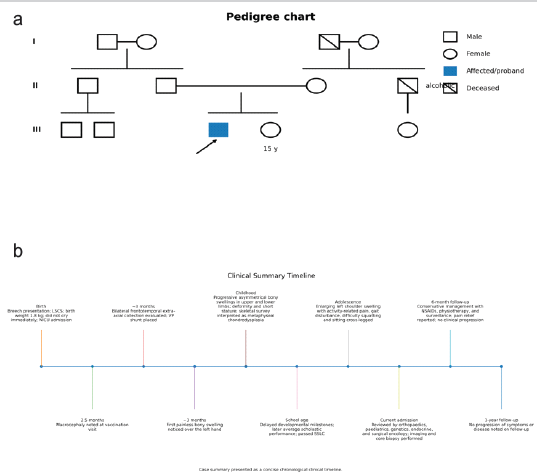
Figure 6: Pedigree and clinical timeline. (a) Simplified pedigree reconstructed from genetics consultation notes showing an unaffected non-consanguineous parental pair, the male proband, and an unaffected sister, with no similarly affected first-degree relatives documented. (b) Clinical timeline showing major perinatal, developmental, musculoskeletal, diagnostic, and follow-up milestones from birth to 1-year post-index management.
Major clinical milestones are summarised in Fig. 6. Genetic testing and advanced cross-sectional imaging were not available for review in the submitted records. As there was no biopsy-proven malignancy, neurovascular compromise, skin breakdown, or radiographic evidence of aggressive soft-tissue destruction, the patient was managed conservatively with non-steroidal anti-inflammatory drugs, physiotherapy, and serial surveillance. At 6 months, he reported pain relief and improved comfort in daily activities. At 1 year, there was no documented clinical progression.
Severe Ollier disease can be difficult to interpret when deformity, disproportionate lesion size, and limited investigations coexist. In this adolescent, the diagnosis was supported by childhood onset, absence of a similar family history, asymmetrical appendicular involvement, and serial radiographs showing multiple expansile intramedullary chondroid lesions with metaphyseal predominance. The literature identified through a PubMed/Medline search using ‘Ollier disease’, ‘enchondromatosis’, ‘proximal humerus’, and ‘chondrosarcoma’ consists largely of case reports, case series, and reviews; therefore, management decisions remain individualised and depend heavily on clinicoradiological correlation [6,7,8]. The feature that made this case clinically important was the giant, enlarging proximal humeral lesion in a patient with widespread skeletal involvement. Its size and shoulder-girdle location raised an appropriate concern for secondary chondrosarcoma, but the available radiographs did not show definite permeative destruction or a soft-tissue mass. The associated short stature, dysmorphic facies, developmental delay, and previous ventriculoperitoneal shunting widened the differential diagnosis. Hereditary multiple osteochondromas were less likely because the lesions were intramedullary rather than exophytic, and Maffucci syndrome was less likely because cutaneous or soft-tissue haemangiomas were not documented. The central clinical question was whether the proximal humeral lesion represented malignant transformation. Painful rapid enlargement, cortical destruction, soft-tissue extension, and lesions in high-risk sites, such as the pelvis and shoulder girdle, are recognised warning signs in Ollier disease [3,4,5,9,10,11]. In this patient, the lesion size and interval enlargement justified surgical oncology review and image-guided biopsy. Histology from the core biopsy showed benign hyaline cartilage without cytological atypia or mitotic activity, which was reassuring. However, a benign core biopsy cannot fully exclude focal malignant change in a large heterogeneous cartilaginous tumour, so the biopsy result was interpreted together with the clinical course and available imaging rather than in isolation. Treatment was planned around symptoms, function, risk of malignancy, and available resources. Major reconstructive or ablative surgery at presentation was not chosen because there was no biopsy-proven malignancy, neurovascular compromise, skin breakdown, or clear radiographic soft-tissue invasion. A conservative plan with analgesia, physiotherapy, and structured surveillance was therefore reasonable, and the patient reported symptomatic improvement at 6 months with no documented clinical progression at 1 year. Follow-up was specifically directed toward early warning features, including persistent or night pain, rapid increase in swelling, cortical breach, soft-tissue extension, neurological symptoms, skin compromise, or pathological fracture.
Limitations:
This report describes a single patient and cannot be generalised to all patients with Ollier disease. Molecular confirmation with IDH1/IDH2 testing could not be performed because of financial constraints. Magnetic resonance imaging (MRI)/computed tomography (CT) of the giant proximal humeral lesion and whole-body mapping with whole-body MRI, positron emission tomography-CT, or bone scintigraphy were not available; consequently, cortical breach, marrow involvement, soft-tissue extension, occult lesions, and total disease burden could not be assessed comprehensively. Follow-up was limited to 1 year, which is insufficient for evaluating lifelong progression or malignant transformation. Serial imaging measurements of lesion volume, growth rate, and objective progression were not systematically recorded. Histology was based on three core biopsy samples rather than complete excision, and conservative management prevented whole-tumour assessment; hence, sampling error remains possible. Standardised functional outcome tools, including the Musculoskeletal Tumour Society score, Disabilities of the Arm, Shoulder, and Hand score, paediatric outcomes data collection instrument, quality-of-life scales, and formal instrumented gait analysis, were not used. Comprehensive endocrine, neurodevelopmental, and cognitive assessments were also unavailable despite short stature, dysmorphic facies, developmental delay, and previous ventriculoperitoneal shunting, leaving some diagnostic overlap with syndromic or genetic conditions incompletely explored. The management reflects a single-centre experience and may not be reproducible in all settings. Most importantly, the absence of malignancy on the index biopsy does not remove the future risk of sarcomatous transformation, making vigilant long-term surveillance essential.
Severe childhood-onset enchondromatosis consistent with Ollier disease may produce dramatic deformity and large lesions that clinically simulate malignant transformation. Longitudinal imaging review, multidisciplinary assessment, selective biopsy, and vigilant surveillance are central to management. When molecular testing or major reconstruction is not feasible, and malignancy is not proven, conservative management may provide symptomatic benefit while maintaining oncological vigilance.
In severe Ollier disease, rapid enlargement of a giant lesion should not automatically be equated with malignancy. Management should be guided by integrated clinicoradiological assessment, selective biopsy of suspicious lesions, and long-term surveillance tailored to available resources.
References
- 1. Silve C, Jüppner H. Ollier disease. Orphanet J Rare Dis 2006;1:37. [Google Scholar] [PubMed]
- 2. Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet 2011;43:1262-5. [Google Scholar] [PubMed]
- 3. Verdegaal SH, Bovée JV, Pansuriya TC, Grimer RJ, Ozger H, Jutte PC, et al. Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: An international multicentre study of 161 patients. Oncologist 2011;16:1771-9. [Google Scholar] [PubMed]
- 4. Kumar A, Jain VK, Bharadwaj M, Arya RK. Ollier disease: Pathogenesis, diagnosis, and management. Orthopaedics 2015;38:e497-506. [Google Scholar] [PubMed]
- 5. El Abiad JM, Robbins SM, Cohen B, Levin AS, Valle DL, Morris CD, et al. Natural history of Ollier disease and Maffucci syndrome: Patient survey and review of clinical literature. Am J Med Genet A 2020;182:1093-103. [Google Scholar] [PubMed]
- 6. Markevičiūtė V, Markevičiūtė MŠ, Stravinskas M. Ollier disease: A case series and literature review. Acta Med Litu 2021;28:181-8. [Google Scholar] [PubMed]
- 7. El Mandour J, Khouchoua S, Adjou N, El Haddad S, Allali N, Chat L. Ollier disease: A case report and literature review. Radiol Case Rep 2023;18:3652-6. [Google Scholar] [PubMed]
- 8. Kramer HD, Valentine MJ, Pettinelli N, Kim J, Kramer RC. Ollier disease: A case report and review of treatment options. Cureus 2023;15:e43816. [Google Scholar] [PubMed]
- 9. Lau JH, Ng KK, Wong WC, Kung BT. Multiple enchondromas in Ollier’s disease: A case report. Radiol Case Rep 2024;19:5033-7. [Google Scholar] [PubMed]
- 10. Kao WL, Wu CL, Lin HY, Wu HT, Chiou HJ [1.1]. Ollier disease with malignant transformation into chondrosarcoma: Case reports and a review of literature. J Radiol Sci 2024;49:134-8. [Google Scholar] [PubMed]
- 11. Michaeli O, Kim SY, Mitchell SG, Jongmans MC, Wasserman JD, Perrino MR, et al. Update on cancer screening in children with syndromes of bone lesions, hereditary leiomyoma and renal cell carcinoma syndrome, and other rare syndromes. Clin Cancer Res 2025;31:457-65. [Google Scholar] [PubMed]






Related Articles in Journal of Orthopaedic Case Reports
 August 1, 2026 Thumb Distal Phalanx Enchondroma: Expanding the Surgical Corridor Through a Modified Volar Lateral Technique – A Case Report
August 1, 2026 Thumb Distal Phalanx Enchondroma: Expanding the Surgical Corridor Through a Modified Volar Lateral Technique – A Case Report July 1, 2026 Diagnostic Dilemma in a Distal Femoral Intramedullary Lesion: Enchondroma Mimicking Low-Grade Chondrosarcoma with Discordant Biopsy Findings: A Case Report
July 1, 2026 Diagnostic Dilemma in a Distal Femoral Intramedullary Lesion: Enchondroma Mimicking Low-Grade Chondrosarcoma with Discordant Biopsy Findings: A Case Report July 1, 2026 Aneurysmal Bone Cyst of the Proximal Humerus Managed with En Bloc Resection, Fibular Strut Grafting, and PHILOS Fixation: A Case Report
July 1, 2026 Aneurysmal Bone Cyst of the Proximal Humerus Managed with En Bloc Resection, Fibular Strut Grafting, and PHILOS Fixation: A Case Report July 1, 2026 Resolution of Proximal Humerus Enchondroma in a 53-Year-Old Female with Curettage and Bone Substitution: A Case Report
July 1, 2026 Resolution of Proximal Humerus Enchondroma in a 53-Year-Old Female with Curettage and Bone Substitution: A Case Report